



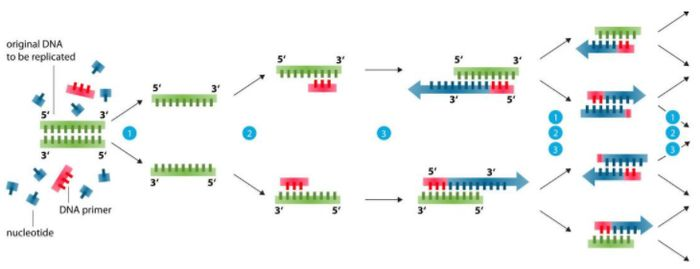
Base de deseño de imprimación (o 99% dos problemas pódense resolver)
1. Lonxitude de cartilla: o libro de texto require 15-30 pb, normalmente uns 20 pb.A condición real é mellor que sexa 18-24 pb para garantir a especificidade, pero canto máis tempo mellor, o cebador demasiado longo tamén reducirá a especificidade e reducirá o rendemento.
2. Intervalo de amplificación do cebador: 200-500 pb é apropiado e o fragmento pódese ampliar a 10 kb en condicións específicas.
3. Base de imprimación: o contido de G + C debe ser do 40-60%, o efecto de amplificación de G + C moi pouco non é bo, demasiado G + C é fácil de aparecer bandas non específicas.O ATGC distribúese mellor de forma aleatoria, evitando agrupacións de máis de 5 nucleótidos de purina ou pirimidina.Multi-gc para o extremo 5′ e secuencias intermedias para aumentar a estabilidade, evite GC rico no extremo 3′, sen GC para as 3 últimas bases ou sen GC para 3 das últimas 5 bases.
4. Evite a estrutura secundaria nos cebadores e evite a complementación entre dous cebadores, especialmente a complementación no extremo 3', se non, formarase un dímero de cebador e xeraranse bandas amplificadas non específicas.
5. As bases no extremo 3' dos cebadores, especialmente a última e a penúltima bases, deberían emparellarse estritamente para evitar o fallo da PCR debido a bases terminais desapareadas.
6. Os cebadores teñen ou poden engadirse con sitios de escisión apropiados, e a secuencia diana amplificada debería ter preferiblemente sitios de escisión adecuados, o que é moi beneficioso para a análise de escisión ou a clonación molecular.
7. Especificidade dos cebadores: os cebadores non deben ter homoloxía evidente con outras secuencias da base de datos de secuencias de ácidos nucleicos.
8. Aprende a usar software: PP5, Oligo6, DNAstar, Vector NTI, primer3 (Este deseño en liña funciona mellor).
O contido anterior pode resolver polo menos o 99% dos problemas de deseño de imprimacións.
Controla os detalles do deseño da imprimación
1. Lonxitude de imprimación
A lonxitude xeral do imprimador é de 18 ~ 30 bases.En xeral, o factor máis importante que determina a temperatura de recocido do cebador é a lonxitude do cebador.A temperatura de recocido do cebador é xeralmente seleccionada (valor Tm -5 ℃), e algúns usan directamente o valor Tm.Pódense usar as seguintes fórmulas para calcular aproximadamente a temperatura de recocido dos cebadores.
Cando a lonxitude do cebador é inferior a 20 pb: [4(G+C)+2(A+T)]-5℃
Cando a lonxitude do cebador é superior a 20 pb: 62,3 ℃ + 0,41 ℃ (% GC) -500/longitud-5 ℃
Ademais, moitos programas tamén se poden usar para calcular a temperatura de recocido, o principio de cálculo será diferente, polo que ás veces o valor calculado pode ter un pequeno espazo.Para optimizar as reaccións de PCR, utilízanse os cebadores máis curtos que garanten temperaturas de recocido non inferiores a 54 ℃ para obter a mellor eficiencia e especificidade.
En xeral, a especificidade do cebador aumenta nun factor de catro por cada nucleótido adicional, polo que a lonxitude mínima do cebador para a maioría das aplicacións é de 18 nucleótidos.O límite superior da lonxitude do cebador non é moi importante, principalmente relacionado coa eficiencia da reacción.Debido á entropía, canto máis longo sexa o cebador, menor será a velocidade á que se hibrida para unirse ao ADN diana para formar un molde estable de dobre cadea para que a ADN polimerase se una.
Cando se usa software para deseñar cebadores, a lonxitude dos cebadores pode determinarse pola súa vez polo valor TM, especialmente para cebadores de PCR cuantitativa de fluorescencia, TM=60℃ ou máis debe controlarse.
2.Contido GC
Xeralmente, o contido de G+C nas secuencias de cebadores é do 40% ~ 60%, e o contido de GC e o valor de Tm dun par de cebadores deben coordinarse.Se o cebador ten unha tendencia grave a GC ou AT, pódese engadir a cantidade adecuada de cola A, T ou G e C ao extremo 5' do cebador.
3. Temperatura de recocido
A temperatura de recocido debe ser 5 ℃ inferior á temperatura de desencadeamento.Se o número de bases de cebadores é pequeno, pódese aumentar a temperatura de recocido adecuadamente, o que pode aumentar a especificidade da PCR.Se o número de bases é grande, a temperatura de recocido pódese reducir adecuadamente.A diferenza de temperatura de recocido entre un par de cebadores de 4℃ ~ 6℃ non afectará o rendemento da PCR, pero idealmente a temperatura de recocido dun par de cebadores é a mesma, que pode variar entre 55℃ e 75℃.
4. Evite a zona de estrutura secundaria da plantilla de amplificación
O mellor é evitar a rexión da estrutura secundaria do modelo ao seleccionar o fragmento amplificado.A estrutura secundaria estable do fragmento obxectivo pódese predecir e estimar mediante o software informático relevante, o que é útil para a selección de modelos.Os resultados experimentais mostran que a expansión a miúdo non ten éxito cando a enerxía libre (△G) da rexión a expandir é inferior a 58,6 lkJ/mol.
5. Incompatibilidade co ADN diana
Cando a secuencia de ADN diana amplificada é grande, un cebador pode unirse a varias partes do ADN diana, dando lugar a que aparezan múltiples bandas no resultado.Nesta ocasión é necesario utilizar a web de probas do software BLAST:http://www.ncbi.nlm.nih.gov/BLAST/.Seleccione Aliñar dúas secuencias (bl2seq).
Pegar as secuencias de cebadores na zona 1 e as secuencias de ADN diana na zona 2 é intercambiable, e BLAST calcula posibilidades complementarias, antisentido e outras, polo que os usuarios non precisan notar se ambas cadeas son cadeas de sentido.Tamén podes introducir o número GI se coñeces o número GI da secuencia na base de datos, polo que non tes que pegar unha sección grande da secuencia.Finalmente, fai clic en Aliñar en 3 para ver se o cebador ten varios sitios homólogos no ADN obxectivo.
6. Terminal de imprimación
O extremo de 3' do cebador é onde comeza a extensión, polo que é importante evitar que os desaxustes comecen alí.O extremo 3' non debe ser máis de 3 G ou C consecutivos, porque isto fará que o cebador se dispare por erro na rexión da secuencia de enriquecemento G+C.O extremo 3' non pode formar ningunha estrutura secundaria, agás nas reaccións especiais de PCR (AS-PCR), o extremo 3' do cebador non se pode combinar.Por exemplo, se a rexión codificante se amplifica, o extremo 3' do cebador non debería terminar na terceira posición do codón, porque a terceira posición do codón é propensa a dexenerarse, o que afectará á especificidade e á eficiencia da amplificación.Cando use cebadores de anexión, consulte a táboa de uso de codóns, preste atención á preferencia biolóxica, non use cebadores de anexión no extremo 3′ e use unha concentración máis alta de cebadores (1uM-3uM).
7. Estrutura secundaria dos cebadores
Os propios cebadores non deberían ter secuencias complementarias, se non, os propios cebadores pregaranse en estruturas de horquilla, e esta estrutura secundaria afectará á unión de cebadores e modelos debido ao impedimento estérico.Se se usa criterio artificial, as bases complementarias continuas dos propios cebadores non deben ser superiores a 3 pb.Non debe haber complementariedade entre os dous cebadores, especialmente o solapamento complementario do extremo 3' debe evitarse para evitar a formación de dímeros de cebadores.En xeral, non debe haber máis de 4 bases consecutivas de homoloxía ou complementariedade entre un par de cebadores.
8. Engade marcadores ou loci
O extremo 5' ten pouco efecto sobre a especificidade da amplificación e, polo tanto, pódese modificar sen afectar á especificidade da amplificación.A modificación do extremo 5 do cebador incluíu: engadir un sitio de restrición enzimática;Biotina marcada, fluorescencia, digoxina, Eu3+, etc. Introducir secuencias de ADN de unión a proteínas;Introducir sitios de mutación, inserir e perder secuencias de mutación e introducir secuencias promotoras, etc. As bases extra afectarán máis ou menos á eficiencia da amplificación e aumentarán as posibilidades de formación de dímeros de cebador, pero hai que facer algunhas concesións para o seguinte paso.As secuencias adicionais que non existen na secuencia diana, como sitios de restrición e secuencias promotoras, pódense engadir ao extremo 5′ do cebador sen afectar á especificidade.Estas secuencias non están incluídas no cálculo dos valores de Tm do cebador, pero deben ser probadas para a súa complementariedade e estrutura secundaria interna.
9. Subclons
Na maioría das veces, a PCR é só clonación preliminar, e despois necesitamos subclonar o fragmento obxectivo en varios vectores, polo que necesitamos deseñar bases adicionais para a seguinte operación no paso da PCR.
A continuación resúmense algunhas secuencias deseñadas para a subclonación.
Engadiuse o sitio de restrición da endonuclease de restrición
Engadir sitios de restrición enzimática é o método máis utilizado para subclonar produtos de PCR.Xeralmente, o sitio de clivaxe é de seis bases, ademais do 5 'final do sitio de clivaxe precisa engadir 2 ~ 3 bases protectoras.Non obstante, o número de bases protectoras requiridas por diferentes encimas é diferente.Por exemplo, SalⅠ non require ningunha base protectora, EcoRⅤ require 1 base protectora, NotⅠ require 2 bases protectoras e Hind Ⅲ require 3 bases protectoras.
LIC engade o rabo
O nome completo de LIC é Ligation-Independent cloning, un método de clonación inventado por Navogen especificamente para a súa parte do vector pET.O portador de pET preparado polo método LIC ten extremos pegajosos dunha cadea simple de 12-15 bases non complementarios, que complementan os extremos adhesivos correspondentes no fragmento de inserción de destino.Para fins de amplificación, a secuencia do cebador 5′ do fragmento inserido debería complementar o vector LIC.A actividade de extracción 3′→5′ da ADN polimerase T4 pode formar un extremo pegajoso dunha única cadea no fragmento inserido despois de pouco tempo.Dado que o produto só se pode formar a partir do recocido mutuo do fragmento de inserción preparado e do vector, este método é moi rápido e eficiente e é unha clonación dirixida.
O clon de TA dirixido engade cola
A clonación TA non foi capaz de dirixir o fragmento a un vector, polo que máis tarde Invitrogen introduciu un vector que podería dirixirse á clonación, que contiña catro GTGGS de bases prominentes nun extremo.Polo tanto, no deseño dos cebadores da PCR, deberían engadirse secuencias complementarias en consecuencia, para que os fragmentos poidan "orientarse".
Se tes pouco tempo, podes probar a síntese directa, combinando o xene co vector, que é o que chamamos síntese do xene ET nos musculadores.
D. Método de clonación In-Fusion
Non se precisa ligase, non se precisa unha reacción longa.Sempre que se introduza unha secuencia nos dous extremos do vector linealizado No deseño dos cebadores, a continuación, o produto da PCR e o vector linealizado engádense á solución enzimática de fusión que contén BSA e colócanse a temperatura ambiente durante media hora, pódese realizar a transformación.Este método é especialmente axeitado para a conversión de grandes volumes.
10. Unir cebador
Ás veces, só se coñece información de secuencia limitada sobre o deseño de cebadores.Por exemplo, se só se coñece a secuencia de aminoácidos, pódese deseñar o cebador de fusión.Un cebador de fusión é unha mestura de diferentes secuencias que representan todas as diferentes posibilidades de bases que codifican un só aminoácido.Para aumentar a especificidade, pode consultar a táboa de uso de codóns para reducir a anexión segundo as preferencias de uso base de diferentes organismos.A hipoxantina pódese combinar con todas as bases para reducir a temperatura de recocido da imprimación.Non use as bases anexas no extremo 3′ do cebador porque o recocido das 3 últimas bases no extremo 3′ é suficiente para iniciar a PCR no sitio incorrecto.Utilízanse concentracións de cebadores máis altas (1 μM a 3 μM) porque os cebadores en moitas mesturas de anexión non son específicos do modelo obxectivo.
Materias primas de PCRcontrol
1. Cantidade de imprimación
A concentración de cada cebador é de 0,1 ~ 1 umol ou 10 ~ 100 pmol.É mellor producir o resultado necesario coa menor cantidade de imprimación.A alta concentración de cebador provocará desaxustes e amplificación inespecífica, e aumentará a posibilidade de formar dímeros entre cebadores.
2. Concentración de imprimación
A concentración de cebadores afecta a especificidade.A concentración óptima do cebador é xeralmente entre 0,1 e 0,5 μM.As concentracións de cebador máis altas conducen á amplificación de produtos inespecíficos.
3. Temperatura de recocido da imprimación
Outro parámetro importante para os cebadores é a temperatura de fusión (Tm).Esta é a temperatura na que o 50% dos cebadores e secuencias complementarias se representan como moléculas de ADN de dobre cadea.Requírese Tm para establecer a temperatura de recocido por PCR.Idealmente, a temperatura de recocido é o suficientemente baixa como para garantir un recocido eficaz dos cebadores coa secuencia diana, pero o suficientemente alta como para reducir a unión inespecífica.Temperatura de recocido razoable de 55 ℃ a 70 ℃.A temperatura de recocido é xeralmente 5 ℃ inferior á Tm da imprimación.
Existen varias fórmulas para fixar Tm, que varían moito dependendo da fórmula utilizada e da secuencia dos cebadores.Dado que a maioría das fórmulas proporcionan un valor estimado de Tm, todas as temperaturas de recocido son só un punto de partida.A especificidade pódese mellorar analizando varias reaccións que aumentan progresivamente a temperatura de recocido.Comeza por debaixo da Tm-5 ℃ estimada e aumenta gradualmente a temperatura de recocido nun incremento de 2 ℃.A temperatura de recocido máis alta reducirá a formación de dímeros de imprimación e produtos non específicos.Para obter mellores resultados, os dous cebadores deben ter valores de Tm aproximados.Se a diferenza de Tm dos pares de cebadores é superior a 5 ℃, os cebadores mostrarán un inicio falso significativo ao usar unha temperatura de recocido máis baixa no ciclo.Se os dous cebadores Tm son diferentes, configure a temperatura de recocido en 5℃ máis baixa que a Tm máis baixa.Alternativamente, para aumentar a especificidade, pódense realizar cinco ciclos primeiro a temperaturas de recocido deseñadas para unha Tm máis alta, seguidos dos ciclos restantes a temperaturas de recocido deseñadas para unha Tm máis baixa.Isto permite obter unha copia parcial do modelo de destino en condicións axustadas.
4. Pureza e estabilidade do imprimador
A pureza estándar dos cebadores personalizados é adecuada para a maioría das aplicacións de PCR.A eliminación dos grupos benzoílo e isobutilo mediante a desalación é mínima e, polo tanto, non interfire coa PCR.Algunhas aplicacións requiren purificación para eliminar todas as secuencias que non sexan de lonxitude completa no proceso de síntese.Estas secuencias truncadas ocorren porque a eficiencia da química de síntese de ADN non é do 100%.Este é un proceso circular que utiliza reaccións químicas repetidas a medida que se engade cada base para facer ADN de 3′ a 5′.Pode fallar en calquera dos ciclos.Os cebadores máis longos, especialmente aqueles de máis de 50 bases, teñen unha gran proporción de secuencias truncadas e poden requirir purificación.
O rendemento dos cebadores vese afectado pola eficacia da química sintética e o método de purificación.As empresas biofarmacéuticas, como Cytology e Shengong, usan todas unha unidade de DO mínima para garantir a produción total de oligonucleósidos.Os imprimadores personalizados envíanse en forma de po seco.É mellor redisolver os cebadores en TE para que a concentración final sexa 100μM.O TE é mellor que a auga desionizada porque o pH da auga adoita ser ácido e provocará a hidrólise dos oligonucleósidos.
A estabilidade dos imprimadores depende das condicións de almacenamento.O po seco e as imprimacións disoltas deben almacenarse a -20 ℃.Os cebadores disoltos en TE a concentracións superiores a 10 μM pódense almacenar de forma estable a -20 ℃ durante 6 meses, pero só poden almacenarse a temperatura ambiente (entre 15 ℃ e 30 ℃) durante menos dunha semana.Os imprimadores en po seco pódense almacenar a -20 C durante polo menos 1 ano e a temperatura ambiente (15 C a 30 C) ata 2 meses.
5. Encimas e as súas concentracións
Na actualidade, a ADN polimerase Taq utilizada é basicamente o encima de enxeñaría xénica sintetizado por bacterias coliformes.A cantidade de encima necesaria para catalizar unha reacción de PCR típica é de aproximadamente 2,5 U (refírese ao volume de reacción total de 100 ul).Se a concentración é demasiado alta, pode levar a unha amplificación inespecífica;se a concentración é demasiado baixa, a cantidade de produto sintético reducirase.
6. Calidade e concentración do dNTP
A calidade do dNTP está moi relacionada coa concentración e a eficiencia da amplificación por PCR.O po de dNTP é granular e a súa variabilidade perde a súa actividade biolóxica se se almacena de forma inadecuada.A solución de dNTP é ácida e debe usarse en alta concentración, cunha solución tampón 1M NaOH ou 1M Tris.HCL para axustar o seu pH a 7,0 ~ 7,5, pequena cantidade de subenvases, almacenamento conxelado a -20 ℃.A conxelación múltiple degradará o dNTP.Na reacción de PCR, o dNTP debe ser de 50 ~ 200 umol/L.Especialmente, debe prestarse atención a que a concentración dos catro DNTPS debe ser igual (preparación de moles igual).Se a concentración dalgún deles é diferente dos demais (maior ou menor), producirase desaxuste.A concentración moi baixa reducirá o rendemento dos produtos de PCR.dNTP pode combinarse con Mg2+ e reducir a concentración de Mg2+ libre.
7. Modelo (xene obxectivo) ácido nucleico
A cantidade e o grao de purificación do ácido nucleico molde é un dos vínculos clave para o éxito ou o fracaso da PCR.Os métodos tradicionais de purificación do ADN adoitan empregar SDS e protease K para dixerir e eliminar os exemplares.As principais funcións do SDS son: disolver lípidos e proteínas na membrana celular, destruíndo así a membrana celular ao disolver proteínas da membrana, e disociar proteínas nucleares na célula, o SDS tamén pode combinarse con proteínas e precipitar;A protease K pode hidrolizar e dixerir proteínas, especialmente as histonas unidas ao ADN, e despois usar fenol e cloroformo disolventes orgánicos para extraer proteínas e outros compoñentes celulares, e usar etanol ou alcohol isopropílico para precipitar o ácido nucleico.O ácido nucleico extraído pódese usar como molde para reaccións de PCR.Para as mostras de detección clínica xeral, pódese usar un método rápido e sinxelo para disolver células, lisar patóxenos, dixerir e eliminar proteínas dos cromosomas para liberar xenes diana e utilizarse directamente para a amplificación por PCR.A extracción de moldes de ARN normalmente usa o método de isotiocianato de guanidina ou protease K para evitar que a RNase degrade o ARN.
8. Concentración de Mg2+
O Mg2+ ten un efecto significativo sobre a especificidade e o rendemento da amplificación por PCR.Na reacción xeral de PCR, cando a concentración de varios dNTP é de 200 umol/L, a concentración adecuada de Mg2+ é de 1,5 ~ 2,0 mmol/L.A concentración de Mg2+ é demasiado alta, a especificidade da reacción diminúe, prodúcese unha amplificación inespecífica, unha concentración moi baixa reducirá a actividade da ADN polimerase Taq, o que provocará a redución dos produtos da reacción.
Os ións de magnesio afectan a varios aspectos da PCR, como a actividade da ADN polimerase, que afecta o rendemento;Outro exemplo é o recocido de cebadores, que afecta á especificidade.O dNTP e o modelo únense ao ión magnesio, reducindo a cantidade de ión magnesio libre necesaria para a actividade enzimática.A concentración óptima de ións magnesio varía para os diferentes pares de cebadores e modelos, pero unha concentración inicial típica de PCR con 200 μM dNTP é de 1,5 mM (nota: para a PCR cuantitativa en tempo real, use unha solución de ión magnesio de 3 a 5 mM cunha sonda fluorescente).As concentracións máis altas de ións magnesio libres aumentan o rendemento, pero tamén aumentan a amplificación inespecífica e diminúen a fidelidade.Para determinar a concentración óptima, realizáronse valoracións de ións de magnesio en incrementos de 0,5 mM de 1 mM a 3 mM.Para reducir a dependencia da optimización de ións magnesio, pódese usar Platinum Taq DNA polimerase.A ADN polimerase Taq de platino é capaz de manter a función nun rango máis amplo de concentracións de ións magnesio que a ADN polimerase Taq e, polo tanto, require menos optimización.
9. Aditivos promotores de PCR
A optimización da temperatura de recocido, o deseño do cebador e a concentración de ións magnesio é suficiente para unha amplificación altamente específica da maioría dos moldes;non obstante, algúns modelos, incluídos aqueles con alto contido de GC, requiren medidas adicionais.Os aditivos que afectan a temperatura de fusión do ADN proporcionan outra forma de mellorar a especificidade e o rendemento do produto.É necesaria a desnaturalización completa do modelo para obter os mellores resultados.
Ademais, a estrutura secundaria impide a unión do cebador e a extensión do encima.
Os aditivos de PCR, incluíndo formamida, DMSO, glicerina, betaína e PCRx Enhancer Solution, melloran a amplificación.O seu posible mecanismo é reducir a temperatura de fusión, axudando así ao recocido dos cebadores e axudando á extensión da ADN polimerase a través da rexión da estrutura secundaria.A solución PCRx ten outras vantaxes.Requírese unha optimización mínima de ións magnesio cando se usa con ADN polimerase Platinum Taq e ADN polimerase Platinum Pfx.Así, a técnica Platinum combínase co aditivo para aumentar a especificidade mentres se reduce a dependencia do terceiro enfoque, a optimización do ión magnesio.Para obter mellores resultados, debe optimizarse a concentración de aditivos, especialmente DMSO, formamida e glicerol, que inhiben a ADN polimerase Taq.

10. Arranque en quente
A PCR de inicio en quente é un dos métodos máis importantes para mellorar a especificidade da PCR ademais dun bo deseño de cebadores.Aínda que a temperatura de elongación óptima da ADN polimerase Taq é de 72 ℃, a polimerase permanece activa a temperatura ambiente.Así, prodúcense produtos inespecíficos cando a temperatura de mantemento é inferior á temperatura de recocido durante a preparación da reacción de PCR e ao comezo do ciclo térmico.Unha vez formados, estes produtos inespecíficos son efectivamente amplificados.A PCR de inicio en quente é particularmente eficaz cando os sitios utilizados para o deseño de cebadores están limitados pola localización de elementos xenéticos, como mutacións dirixidas ao sitio, clonación de expresións ou construción e manipulación de elementos xenéticos utilizados para a enxeñaría do ADN.
Un método común para limitar a actividade da ADN polimerase Taq é preparar a solución de reacción de PCR sobre xeo e colocala nun aparello de PCR prequecido.Este método é sinxelo e barato, pero non completa a actividade do encima e, polo tanto, non elimina completamente a amplificación de produtos non específicos.
O cebado térmico atrasa a síntese de ADN ao inhibir un compoñente esencial ata que o aparello de PCR alcanza a temperatura de desnaturalización.A maioría dos métodos manuais de iniciación térmica, incluíndo a adición retardada da ADN polimerase Taq, son complicados, especialmente para aplicacións de alto rendemento.Outros métodos de imprimación térmica usan un escudo de cera para encerrar un compoñente esencial, incluíndo ións de magnesio ou encimas, ou para illar fisicamente compoñentes reactivos, como moldes e tampones.Durante o ciclo térmico, os distintos compoñentes son liberados e mestúranse xuntos mentres a cera se funde.Do mesmo xeito que o método de inicio manual en quente, o método de protección contra cera é engorroso e propenso á contaminación e non é axeitado para aplicacións de alto rendemento.
A ADN polimerase de platino é conveniente e eficiente para a PCR de inicio automático en quente.A ADN polimerase Taq de platino está formada por ADN polimerase Taq recombinante combinada cun anticorpo monoclonal contra a ADN polimerase Taq.Os anticorpos son formulados por PCR para inhibir a actividade enzimática durante un período prolongado de temperatura.A ADN polimerase Taq foi liberada na reacción durante o illamento de 94 ℃ da etapa de desnaturalización, restablecendo a actividade completa da polimerase.En contraste coa ADN polimerase Taq modificada quimicamente para a iniciación térmica, o encima platino non require illamento prolongado a 94 ℃ (10 a 15 minutos) para activar a polimerase.Coa ADN polimerase PlatinumTaq, o 90% da actividade da ADN polimerase Taq restableceuse despois de 2 minutos a 94 ℃.

11. Nest-PCR
As sucesivas roldas de amplificación utilizando cebadores aniñados poden mellorar a especificidade e a sensibilidade.A primeira rolda é unha amplificación estándar de 15 a 20 ciclos.Unha pequena fracción do produto de amplificación inicial diluíuse de 100 a 1000 veces e engadiuse á segunda rolda de amplificación durante 15 a 20 ciclos.Alternativamente, o produto amplificado inicial pódese dimensionar mediante purificación en xel.Na segunda rolda de amplificación úsase un cebador aniñado, que pode unirse á secuencia diana dentro do primeiro cebador.O uso da PCR aniñada reduce a posibilidade de amplificación de múltiples sitios diana porque hai poucas secuencias diana complementarias a ambos os conxuntos de cebadores.O mesmo número total de ciclos (30 a 40) cos mesmos cebadores amplificaron os sitios inespecíficos.A PCR anidada aumenta a sensibilidade de secuencias diana limitadas (por exemplo, mrnas raras) e mellora a especificidade das PCRS difíciles (por exemplo, 5′ RACE).
12. PCR descendente
A PCR descendente mellora a especificidade empregando condicións de recocido axustado durante os primeiros ciclos de PCR.O ciclo comeza a unha temperatura de recocido aproximadamente 5℃ superior á Tm estimada, despois cada ciclo redúcese de 1℃ a 2℃ ata que a temperatura de recocido sexa inferior a Tm 5℃.Só se amplificará o modelo de destino con maior homoloxía.Estes produtos seguen a expandirse en ciclos posteriores, eliminando os produtos inespecíficos amplificados.A PCR descendente é útil para métodos nos que non se coñece o grao de homoloxía entre o cebador e a plantilla diana, como a pegada dixital de ADN AFLP.
Kits de PCR relacionados
O 2× PCR HeroTMO sistema Mix ten unha maior tolerancia aos inhibidores da PCR que o sistema PCR Mix ordinario e pode xestionar facilmente a amplificación por PCR de varios modelos complexos.O sistema de reacción único e Taq Hero de alta eficiencia fan que a reacción PCR teña unha maior eficiencia de amplificación, especificidade e sensibilidade.
Maior eficiencia de amplificación
Ten actividade de ADN polimerase 5'→3' e actividade exonuclease 5'→3', sen actividade exonuclease 3'→5'.
Real Time PCR Easyᵀᴹ-SYBR Green I Kit
Específico: o tampón optimizado e o encima Taq de inicio en quente poden evitar a amplificación inespecífica e a formación de dímeros de cebador
Alta sensibilidade: pode detectar copias baixas do modelo
O kit usa un único reactivo de transcrición inversa Foregene e Foregene HotStar Taq DNA Polymerase combinados cun sistema de reacción único para mellorar eficazmente a eficiencia e especificidade da amplificación da reacción.
Hora de publicación: maio-09-2023



