



Como un novo no laboratorio, non é un bo traballo eliminar as plantas positivas dun grupo de plantas cunha taxa de conversión baixa.En primeiro lugar, hai que extraer o ADN dunha gran cantidade de mostras unha por unha e despois detectaranse os xenes estraños mediante PCR.Non obstante, os resultados adoitan ser espazos en branco e bandas con algúns elementos ocasionalmente, pero é imposible determinar se hai deteccións perdidas ou falsas..É moi impotente afrontar un proceso e resultados tan experimentais?Non te preocupes, o irmán ensínache a eliminar as plantas positivas transxénicas con facilidade e precisión.
Paso 1
Deseñar cebadores de detección

Determine o xene endóxeno e o xene exóxeno que se vai detectar segundo a mostra que se vai probar e seleccione unha secuencia representativa de 100-500 pb no xene para o deseño do cebador.Os bos cebadores poden garantir a precisión dos resultados da detección e acurtar o tempo de detección (consulte o apéndice para os cebadores de detección de uso habitual).
Aviso: os cebadores recentemente deseñados deben optimizar as condicións de reacción e verificar a precisión, a precisión e o límite de detección da detección antes da detección a gran escala.
Paso 2
Deseño protocolo experimental

Control positivo: use o ADN purificado que contén o fragmento diana como molde para determinar se o sistema de reacción de PCR e as condicións son normais.
Control negativo/en branco: use o molde de ADN ou ddH2O que non conteña o fragmento diana como molde para detectar se existe unha fonte de contaminación no sistema de PCR.
Control interno de referencia: use a combinación cebador/sonda do xene endóxeno da mostra que se vai probar para avaliar se o molde pode detectarse mediante PCR.
Aviso:
Deben establecerse controis positivos, negativos/en branco e controis de control interno para cada proba para avaliar a validez dos resultados experimentais.
Preparación do experimento

Antes do uso, observe se a solución se mestura uniformemente.Se se atopa precipitación, debe disolverse e mesturarse segundo as instrucións antes do uso.A mestura de 2×PCR debe pipetearse e mesturarse repetidamente cunha micropipeta antes do uso para evitar unha distribución desigual de ións.
Aviso:
Saca o manual e léao atentamente, e fai os preparativos antes do experimento de acordo estrito cos requisitos do manual.
Paso 4
Preparar o sistema de reacción PCR

Segundo o protocolo experimental, mestura os cebadores, H2O e 2×PCR mestura uniformemente, centrifuga e distribúeos en cada tubo de reacción.
Aviso:
Para probas a gran escala ou a longo prazo, recoméndase utilizar un sistema de reacción de PCR que conteña encima UNG, que pode evitar eficazmente a contaminación dos aerosols causada polos produtos de PCR.
Paso 5
Engadir modelo de reacción

Usando a tecnoloxía Direct PCR, non é necesario un proceso tedioso de purificación de ácido nucleico, o modelo de mostra pódese preparar en 10 minutos e pódese engadir o sistema de reacción PCR correspondente.
Aviso:
O método de escisión ten un mellor efecto de detección e o produto obtido pódese usar para múltiples reaccións de detección.

5.1: Expansión directa das follas
Segundo o tamaño da imaxe do manual, corte o tecido da folla cun diámetro de 2-3 mm e colócao no sistema de reacción PCR.
Nota: Asegúrese de que os fragmentos de follas estean completamente inmersos na solución de reacción de PCR e non engadas exceso de tecido foliar.
5.2: Método de división das follas
Cortar o tecido da folla cun diámetro de 5-7 mm e colócao nun tubo de centrífuga.Se escollas follas maduras, evita usar os tecidos da vea principal da folla.Pipete 50ul Buffer P1 lisado nun tubo de centrífuga para asegurarse de que o lisado poida mergullar completamente o tecido da folla, colócase nun termociclador ou nun baño de metal e lise a 95 °C durante 5-10 minutos.

Engade 50ul de solución de neutralización Buffer P2 e mestura ben.O lisado resultante pódese usar como molde e engadirse ao sistema de reacción de PCR.
Nota: a cantidade de modelo está entre o 5-10% do sistema de PCR e non debe exceder o 20% (por exemplo, nun sistema de PCR de 20 μl, engade 1-2 μl de solución de lise, non máis de 4 μl).
Paso 6
Reacción de PCR

Despois de centrifugar o tubo de reacción de PCR, colócase nun instrumento de PCR para amplificación.
Aviso:
A reacción usa un molde non purificado para a amplificación, polo que o número de ciclos de amplificación é de 5-10 ciclos máis que cando se usa un molde de ADN purificado.
Paso 7
Detección por electroforese e análise de resultados
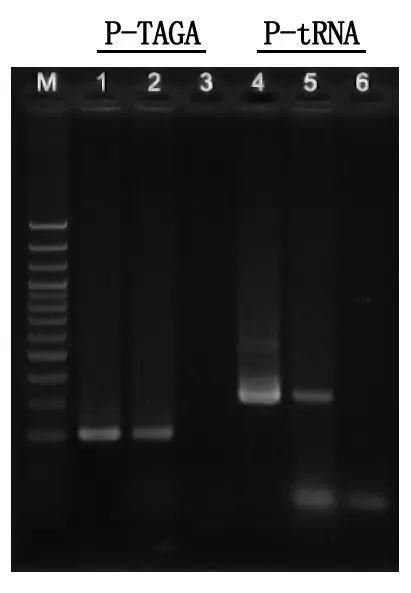
M: Escaleira de ADN de 100 pb
1\4: Método do ADN purificado
2\5: Método de PCR directa
3\6: control en branco
QC:
Os resultados das probas dos distintos controis establecidos no experimento deben cumprir as seguintes condicións.En caso contrario, debe analizarse a causa do problema e realizar a proba de novo despois de que se elimine o problema.
Táboa 1. Resultados normais das probas de varios grupos control
*Cando se usa o plásmido como control positivo, o resultado da proba do xene endóxeno pode ser negativo
Xuízo de resultado:
A. O resultado da proba do xene endóxeno da mostra é negativo, o que indica que o ADN axeitado para a detección ordinaria da PCR non se pode extraer da mostra ou que o ADN extraído contén inhibidores da reacción de PCR, e o ADN debe extraerse de novo.
B. O resultado da proba do xene endóxeno da mostra é positivo e o resultado da proba do xene exóxeno é negativo, o que indica que a mostra extrae ADN axeitado para a detección por PCR ordinaria e pódese xulgar que o xene XXX non se detecta na mostra.
C. O resultado da proba do xene endóxeno da mostra é positivo e o resultado da proba do xene exóxeno é positivo, o que indica que se extraeu da mostra ADN axeitado para a detección por PCR ordinaria e que o ADN da mostra contén o xene XXX.Pódense realizar máis experimentos de confirmación.
Paso 8
Deseñar cebadores de detección

Despois do experimento, use unha solución de hipoclorito de sodio ao 2% e unha solución de etanol ao 70% para limpar a área experimental para evitar a contaminación ambiental.
Hora de publicación: 08-09-2021



