



RT-qPCR desenvólvese a partir da tecnoloxía PCR común.Engade produtos químicos fluorescentes (colorantes fluorescentes ou sondas fluorescentes) ao sistema de reacción tradicional da PCR, e detecta o proceso de recocido e extensión da PCR en tempo real segundo os seus diferentes mecanismos luminiscentes.Os cambios de sinal fluorescente no medio utilízanse para calcular a cantidade de cambio de produto en cada ciclo de PCR.Actualmente, os métodos máis comúns son o método de tintura fluorescente e o método de sonda.
Método de tintura fluorescente:
Algúns colorantes fluorescentes, como SYBR Green Ⅰ, PicoGreen, BEBO, etc., non emiten luz por si mesmos, pero emiten fluorescencia despois de unirse ao suco menor do dsDNA.Polo tanto, ao comezo da reacción de PCR, a máquina non pode detectar o sinal fluorescente.Cando a reacción pasa á fase de recocido-extensión (método de dúas etapas) ou de extensión (método de tres pasos), as dobres cadeas ábrense neste momento e a nova ADN polimerase Durante a síntese da cadea, as moléculas fluorescentes combínanse no suco menor do dsDNA e emiten fluorescencia.A medida que aumenta o número de ciclos de PCR, máis e máis colorantes combínanse con dsDNA e o sinal fluorescente tamén se mellora continuamente.Tome SYBR Green Ⅰ como exemplo.
Método de sonda:
A sonda Taqman é a sonda de hidrólise máis utilizada.Hai un grupo fluorescente no extremo 5′ da sonda, xeralmente FAM.A sonda en si é unha secuencia complementaria ao xene diana.Hai un grupo de extinción fluorescente no extremo 3′ do fluoróforo.Segundo o principio de transferencia de enerxía de resonancia de fluorescencia (Förster resonance energy transfer, FRET), cando o grupo fluorescente informador (molécula fluorescente doadora) e o grupo fluorescente extintor (molécula fluorescente aceptora) Cando o espectro de excitación se solapa e a distancia é moi próxima (7-10 nm), a excitación da molécula fluorescente do doante pode inducir a autofluorescencia da molécula doante. está debilitado.Polo tanto, ao comezo da reacción de PCR, cando a sonda está libre e intacta no sistema, o grupo fluorescente informador non emitirá fluorescencia.Ao recocer, o cebador e a sonda únense ao molde.Durante a etapa de extensión, a polimerase sintetiza continuamente novas cadeas.A ADN polimerase ten actividade exonuclease 5′-3′.Ao chegar á sonda, a ADN polimerase hidrolizará a sonda do molde, separará o grupo fluorescente informador do grupo fluorescente extintor e liberará o sinal fluorescente.Dado que existe unha relación un a un entre a sonda e a plantilla, o método da sonda é superior ao método da tintura en canto á precisión e sensibilidade da proba.
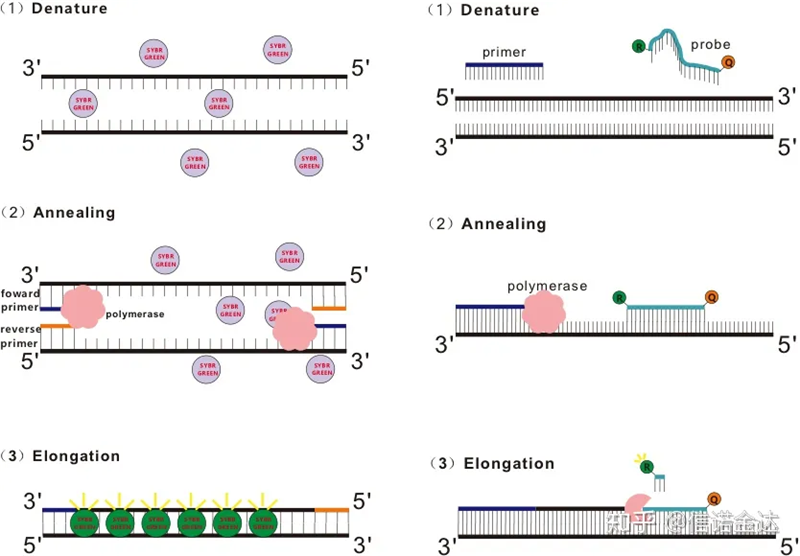
Figura 1 Principio de qRT-PCR
Deseño de imprimación
Principios:
Os cebadores deben deseñarse na rexión conservada da serie de ácidos nucleicos e ter especificidade.
É mellor usar a secuencia de ADNc, e a secuencia de ARNm tamén é aceptable.Se non, descubra o deseño da rexión cds da secuencia de ADN.
A lonxitude do produto cuantitativo fluorescente é de 80-150 pb, a máis longa é de 300 pb, a lonxitude do cebador é xeralmente entre 17-25 bases e a diferenza entre os cebadores augas arriba e augas abaixo non debe ser demasiado grande.
O contido G+C está entre o 40% e o 60%, e o 45-55% é o mellor.
O valor TM está entre 58-62 graos.
Intente evitar dímeros de cebadores e autodímeros, (non aparecen máis de 4 pares de bases complementarias consecutivas) estrutura de horquilla, se é inevitable, faga ΔG<4,5 kJ/mol* Se non pode asegurarse de que se eliminou o ADNg durante a transcrición inversa Limpa, o mellor é deseñar os cebadores do intrón. 3) cebadores e non
específico A homoloxía da secuencia amplificada heteroxénea é preferiblemente inferior ao 70% ou ten 8 homoloxías de bases complementarias.
Base de datos:
CottonFGD busca por palabras clave
Deseño de imprimación:
Deseño de imprimación IDT-qPCR
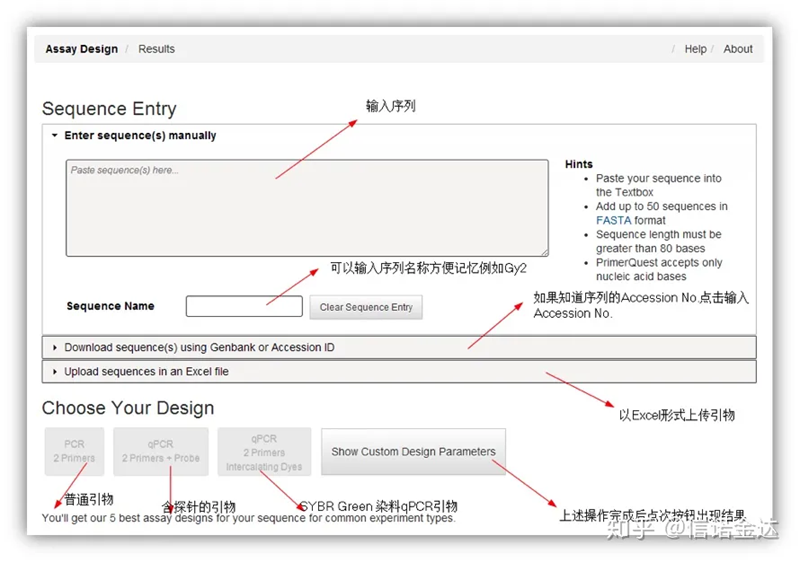
Fig2 Páxina da ferramenta de deseño de cartografía en liña de IDT
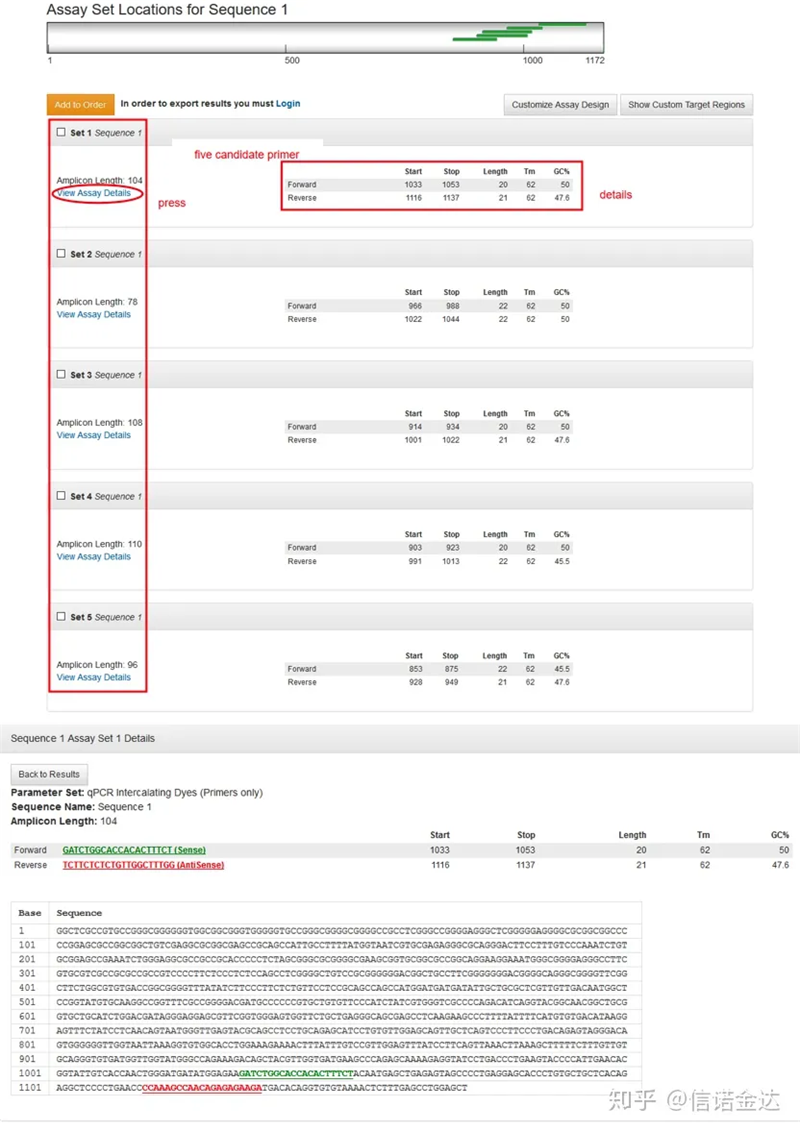
Visualización da páxina de resultados Fig3
Deseño de cebadores de lncRNA:
lncRNA:os mesmos pasos que o ARNm.
miRNA:O principio do método stem-loop: dado que todos os miRNA son secuencias curtas duns 23 nt, non se pode realizar a detección directa por PCR, polo que se utiliza a ferramenta de secuencia stem-loop.A secuencia tronco-bucle é un ADN monocatenario duns 50 nt, que pode formar unha estrutura de horquilla por si só.3 'O extremo pódese deseñar como unha secuencia complementaria ao fragmento parcial de miARN, entón o miARN obxectivo pode conectarse á secuencia de bucle tronco durante a transcrición inversa, e a lonxitude total pode alcanzar os 70 pb, que está en liña coa lonxitude do produto amplificado determinada pola qPCR.Deseño do cebador de miRNA de cola.
Detección específica de amplificación:
Base de datos de explosión en liña: explosión de CottonFGD por semellanza de secuencia
Explosión local: Consulte o uso de Blast+ para facer explosión local, Linux e Macos poden establecer directamente unha base de datos local, o sistema win10 tamén se pode facer despois de instalar Ubuntu bash.Crea unha base de datos de explosión local e explosión local;abrir ubuntu bash en win10.
Aviso: o algodón de montaña e o algodón de illa do mar son cultivos tetraploides, polo que o resultado da explosión adoita ser de dúas ou máis coincidencias.No pasado, o uso de CD NAU como base de datos para realizar explosión é probable que atope dous xenes homólogos con só poucas diferenzas de SNP.Normalmente, os dous xenes homólogos non se poden separar mediante o deseño do cebador, polo que son tratados como iguais.Se hai un indel obvio, o cebador adoita deseñarse no indel, pero isto pode levar á estrutura secundaria do cebador. A enerxía libre aumenta, o que provoca unha diminución da eficiencia de amplificación, pero isto é inevitable.
Detección da estrutura secundaria do cebador:
Pasos:abrir oligo 7 → introducir a secuencia do modelo → pechar a ventá secundaria → gardar → localizar o cebador no modelo, premer ctrl+D para establecer a lonxitude do cebador → analizar varias estruturas secundarias, como corpo de autodimerización, heterodímero, horquilla, desaxuste, etc. As dúas últimas imaxes da Figura 4 son os resultados das probas dos cebadores.O resultado do cebador frontal é bo, non hai unha estrutura obvia de dímero e forquilla, non hai bases complementarias continuas e o valor absoluto da enerxía libre é inferior a 4,5, mentres que o cebador traseiro mostra continuo As 6 bases son complementarias e a enerxía libre é 8,8;ademais aparece un dímero máis grave no extremo 3′, e aparece un dímero de 4 bases consecutivas.Aínda que a enerxía libre non é alta, o dímero 3′ Chl pode afectar seriamente a especificidade e a eficiencia da amplificación.Ademais, é necesario comprobar as horquillas, heterodímeros e desaxustes.
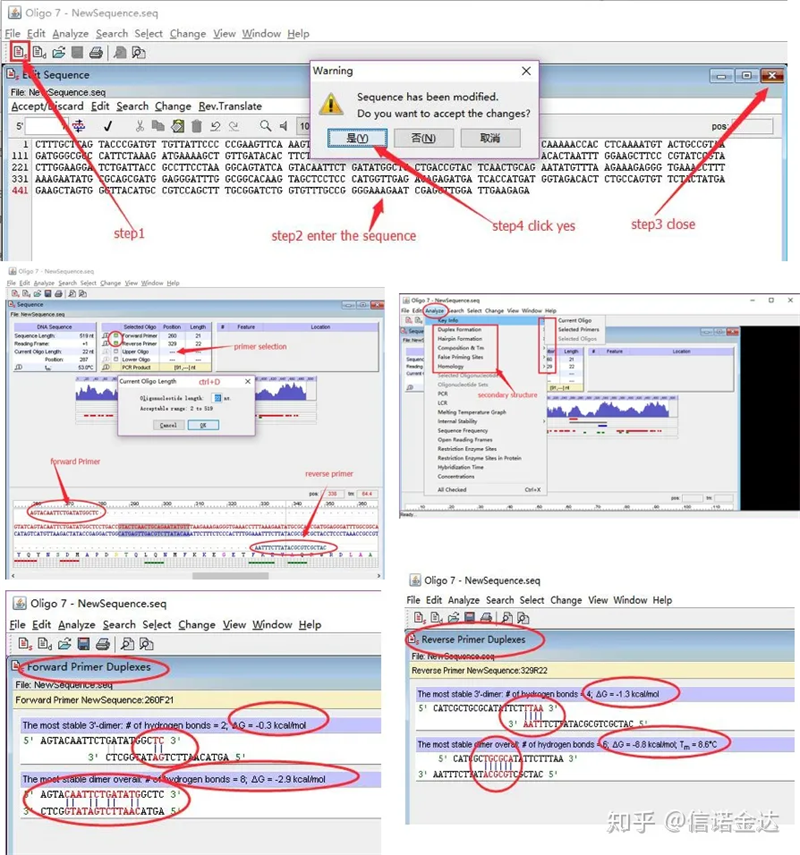
Fig3 resultados da detección de oligo7
Detección de eficiencia de amplificación:
A eficiencia de amplificación da reacción da PCR afecta seriamente aos resultados da PCR.Tamén na qRT-PCR, a eficiencia de amplificación é particularmente importante para os resultados cuantitativos.Eliminar outras substancias, máquinas e protocolos no tampón de reacción.A calidade dos cebadores tamén ten unha gran influencia na eficacia de amplificación da qRT-PCR.Para garantir a precisión dos resultados, tanto a cuantificación de fluorescencia relativa como a cuantificación de fluorescencia absoluta precisan detectar a eficacia de amplificación dos cebadores.Recoñécese que a eficacia de amplificación qRT-PCR efectiva está entre o 85% e o 115%.Hai dous métodos:
1. Método de curva estándar:
a.Mestura cDNA
b.Dilución en gradiente
c.qPCR
d.Ecuación de regresión lineal para calcular a eficiencia de amplificación
2. LinRegPCR
LinRegPCR é un programa para a análise de datos de RT-PCR en tempo real, tamén chamados datos de PCR cuantitativos (qPCR) baseados en SYBR Green ou química similar.O programa usa datos non corrixidos na liña de base, realiza unha corrección de liña base en cada mostra por separado, determina unha xanela de linealidade e despois usa a análise de regresión lineal para axustar unha liña recta a través do conxunto de datos da PCR.A partir da pendente desta liña calcúlase a eficiencia da PCR de cada mostra individual.A eficiencia media da PCR por amplicón e o valor Ct por mostra utilízanse para calcular unha concentración inicial por mostra, expresada en unidades de fluorescencia arbitrarias.A entrada e saída dos datos realízanse a través dunha folla de cálculo de Excel.Só mostra
é necesario mesturar, sen gradiente
son necesarios pasos:(Tome Bole CFX96 como exemplo, non moi Máquina con ABI claro)
experimento:é un experimento estándar de qPCR.
Saída de datos de qPCR:LinRegPCR pode recoñecer dúas formas de ficheiros de saída: RDML ou resultado de amplificación de cuantificación.De feito, é o valor de detección en tempo real do número de ciclo e do sinal de fluorescencia pola máquina, e a amplificación obtense analizando o valor de cambio de fluorescencia da eficiencia do segmento lineal.
Selección de datos: En teoría, o valor RDML debería ser utilizable.Estímase que o problema do meu ordenador é que o software non pode recoñecer RDML, polo que teño o valor de saída de Excel como datos orixinais.Recoméndase realizar unha selección aproximada dos datos primeiro, como a falla de engadir mostras, etc. Os puntos pódense eliminar nos datos de saída (por suposto, non pode borralos, LinRegPCR ignorará estes puntos na fase posterior)
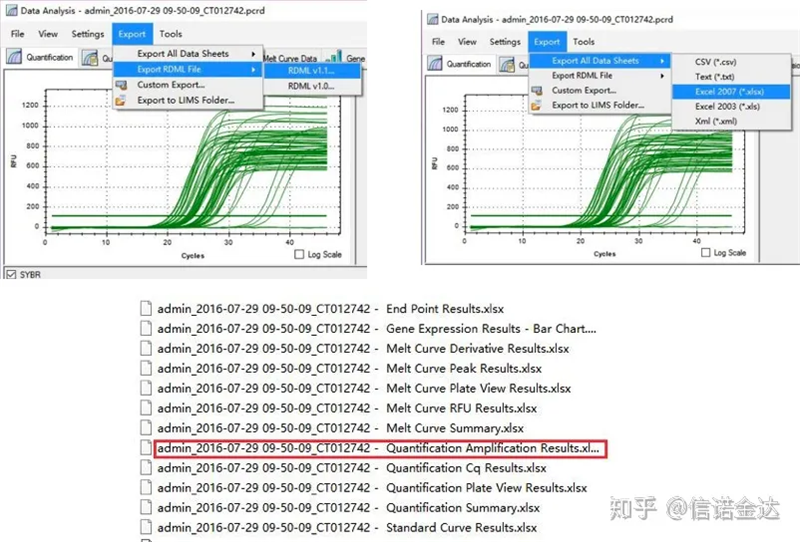
Fig5 Exportación de datos de qPCR
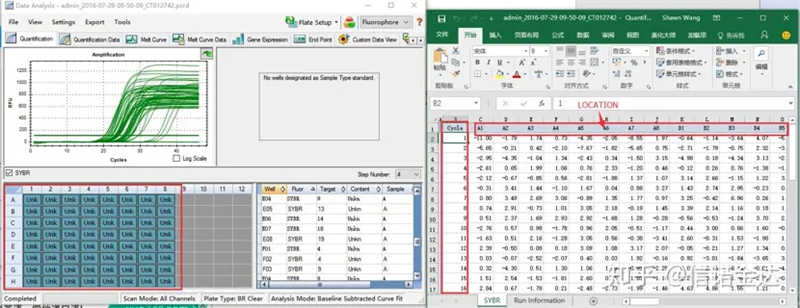
Fig6 selección de mostras candidatas
Entrada de datos:Abre resultados de amplificación de cualificación.xls, → abre LinRegPCR → ficheiro → ler desde Excel → selecciona os parámetros como se mostra na Figura 7 → Aceptar → fai clic en determinar liñas de base
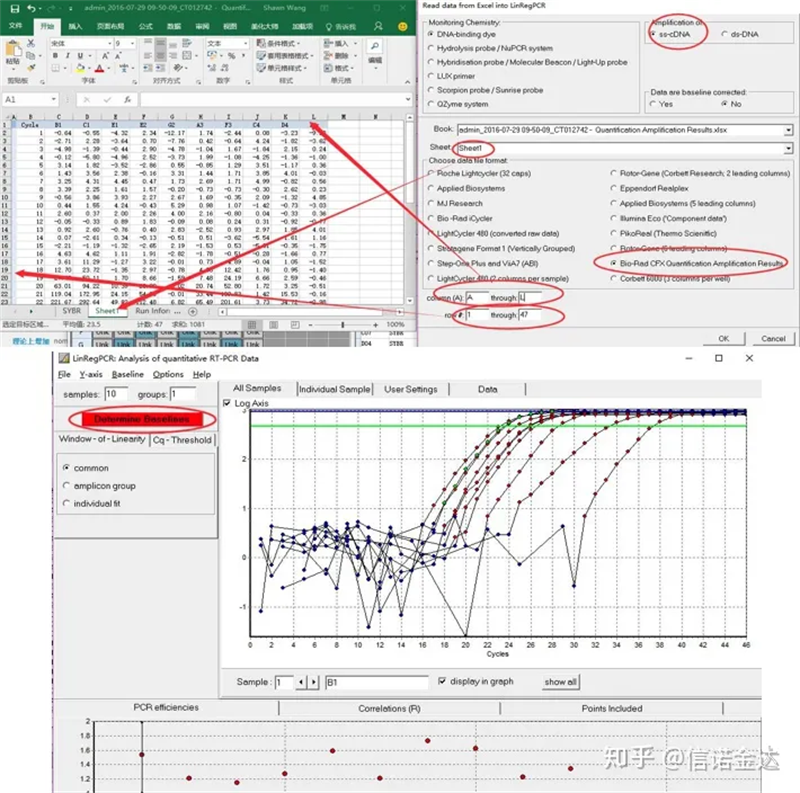
Fig 7 pasos da entrada de datos linRegPCR
Resultado:Se non hai repetición, non é necesario agrupar.Se hai repetición, a agrupación pódese editar na agrupación de mostra e insírese o nome do xene no identificador, e entón o mesmo xene agruparase automaticamente.Finalmente, fai clic no ficheiro, exporta Excel e consulta os resultados.Mostraranse a eficiencia de amplificación e os resultados R2 de cada pozo.En segundo lugar, se se divide en grupos, mostrarase a eficiencia media de amplificación corrixida.Asegúrese de que a eficiencia de amplificación de cada cebador estea entre o 85% e o 115%.Se é demasiado grande ou moi pequeno, significa que a eficiencia de amplificación do cebador é pobre.
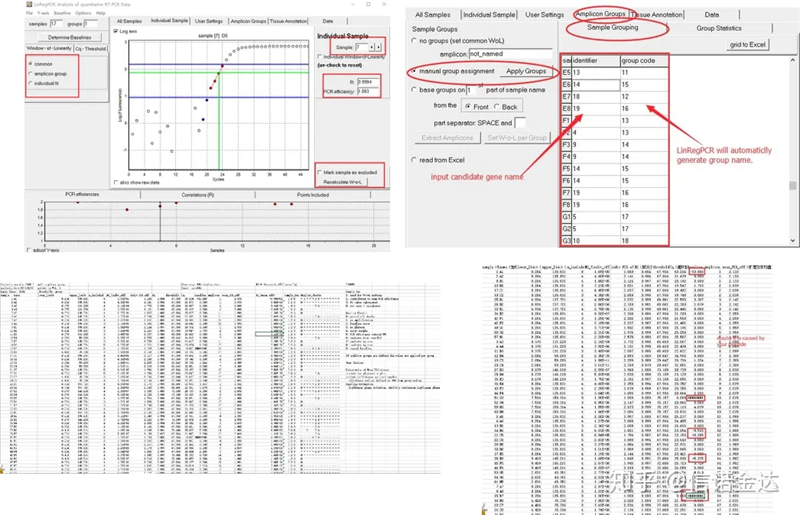
Fig 8 Resultados e saída de datos
Proceso experimental:
Requisitos de calidade do ARN:
Pureza:1.72.0 indica que pode haber isotiocianato residual.O ácido nucleico limpo A260/A230 debería estar en torno a 2. Se hai unha forte absorción a 230 nm, indica que hai compostos orgánicos como os ións fenato.Ademais, pódese detectar mediante electroforese en xel de agarosa ao 1,5%.Apunte o marcador, porque o ARNs non ten desnaturalización e o logaritmo do peso molecular non ten unha relación lineal e o peso molecular non se pode expresar correctamente.Concentración: teoricamentenonmenos de 100 ng/ul, se a concentración é demasiado baixa, a pureza xeralmente é baixa e non alta
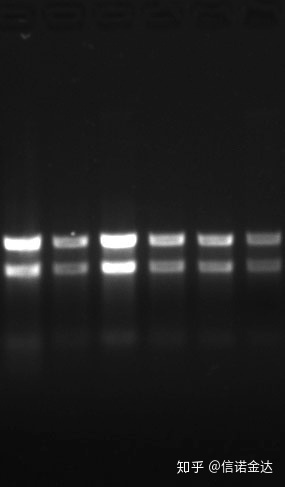
Fig 9 xel de ARN
Ademais, se a mostra é preciosa e a concentración de ARN é alta, recoméndase facer alícuotas despois da extracción e diluír o ARN ata unha concentración final de 100-300 ng/ul para a transcrición inversa.Eno proceso de transcrición inversa, cando se transcribe o ARNm, os cebadores oligo (dt) que poden unirse especificamente ás colas de poliA úsanse para a transcrición inversa, mentres que o lncRNA e o circRNA usan cebadores aleatorios hexámeros (Random 6 mer) para a transcrición inversa do ARN total.Moitas empresas lanzaron agora kits especiais de colas.Para o método de bucle de talo, o método de cola é máis cómodo, de alto rendemento e aforro de reactivos, pero o efecto de distinguir os miARN da mesma familia non debería ser tan bo como o método de bucle de talo.Cada kit de transcrición inversa ten requisitos para a concentración de cebadores específicos de xenes (stem-loops).A referencia interna utilizada para o miARN é U6.No proceso de inversión de bucle de talo, un tubo de U6 debe invertirse por separado e os cebadores frontal e traseiro de U6 deben engadirse directamente.Tanto o circRNA como o lncRNA poden usar HKG como referencia interna.Endetección de cDNA,
se non hai ningún problema co ARN, o cDNA tamén debería estar ben.Non obstante, se se persegue a perfección do experimento, o mellor é utilizar un xene de referencia interno (xene de referencia, RG) que poida distinguir o ADNg dos cds.Xeralmente, RG é un xene doméstico., HKG) como se mostra na Figura 10;Nese momento, estaba facendo proteína de almacenamento de soia e utilizaba actina7 que contén intróns como referencia interna.O tamaño do fragmento amplificado deste cebador no ADNg era de 452 pb, e se se usaba cDNA como molde, era de 142 pb.A continuación, os resultados das probas descubriron que parte do cDNA estaba realmente contaminado por gDNA, e tamén demostraron que non había ningún problema co resultado da transcrición inversa e que podía usarse como modelo para a PCR.É inútil realizar electroforese en xel de agarosa directamente co ADNc, e é unha banda difusa, que non resulta convincente.
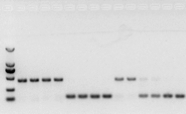
Figura 10 Detección de cDNA
Determinación das condicións de qPCRxeralmente non hai problema segundo o protocolo do kit, principalmente no paso de valor tm.Se algúns cebadores non están ben deseñados durante o deseño do cebador, o que resulta nunha gran diferenza entre o valor tm e os 60°C teóricos, recoméndase que o ADNc Despois de mesturar as mostras, realice unha PCR en gradiente con cebadores e intente evitar establecer a temperatura sen bandas como o valor TM.
Análise de datos
O método de procesamento de PCR cuantitativo de fluorescencia relativa convencional é basicamente segundo 2-ΔΔCT.Modelo de tratamento de datos.
Produtos relacionados:
PCR en tempo real fácilTM -Taqman
PCR en tempo real fácilTM -SYBR VERDE I
RT Easy I (Premestura mestra para a síntese de ADNc da primeira cadea)
RT Easy II (Premestura mestra para a síntese de ADNc da primeira cadea para qPCR)
Hora de publicación: 14-mar-2023



