



Os xenes son as unidades xenéticas básicas que controlan os trazos.Agás os xenes dalgúns virus, que están compostos por ARN, os xenes da maioría dos organismos están compostos por ADN..A maioría das enfermidades dos organismos son causadas pola interacción entre os xenes e o medio ambiente.A terapia xénica pode esencialmente curar ou aliviar moitas enfermidades.A terapia xénica considérase unha revolución no campo da medicina e da farmacia.Os fármacos de terapia xénica nun sentido amplo inclúen fármacos baseados en fármacos de ADN modificado con ADN (como fármacos de terapia xénica in vivo baseados en vectores virais, fármacos de terapia xénica in vitro, fármacos plasmídicos nus, etc.) e fármacos de ARN (como fármacos con oligonucleótidos antisentido, fármacos con ARNm e terapia xénica con ARNm, etc.);sentido estreito Os fármacos de terapia xénica inclúen principalmente fármacos de ADN plasmídico, fármacos de terapia xénica baseados en vectores virais, fármacos de terapia xénica baseados en vectores bacterianos, sistemas de edición de xenes e fármacos de terapia celular para a modificación xénica in vitro.Despois de anos de desenvolvemento tortuoso, os fármacos de terapia xénica lograron resultados clínicos alentadores.(Sen contar as vacinas de ADN e as vacinas de ARNm) Na actualidade, 45 fármacos de terapia xénica foron aprobados para a súa comercialización no mundo.Un total de 9 terapias xénicas foron aprobadas para a súa comercialización este ano, incluíndo 7 terapias xénicas aprobadas para a súa comercialización por primeira vez este ano, a saber: CARVYKTI, Amvuttra, Upstaza, Roctavian, Hemgenix, Adstiladrin e Ebvallo, (Nota: as outras dúas foron aprobadas nos Estados Unidos este ano. foi aprobado pola FDA para a súa comercialización nos Estados Unidos en agosto de 2022 e a Unión Europea en 2019; .) Co lanzamento de cada vez máis produtos de terapia xénica e o rápido desenvolvemento da tecnoloxía de terapia xénica, a terapia xénica está a piques de iniciar un período de rápido desenvolvemento.
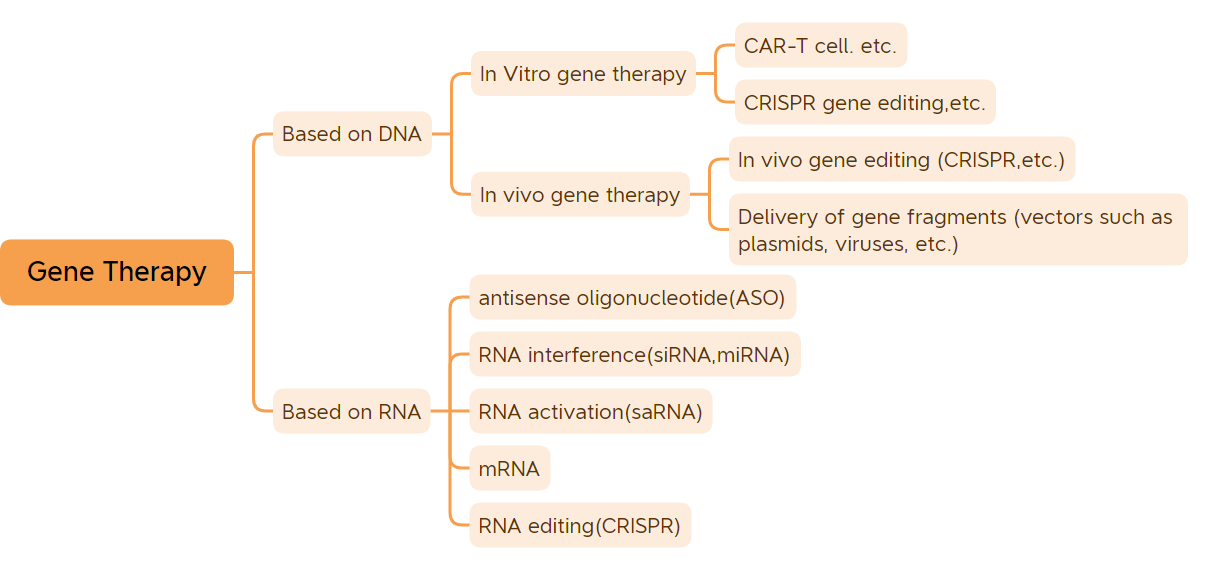
Clasificación da terapia xénica (Fonte da imaxe: Bio-Matrix)
Este artigo enumera 45 terapias xénicas (excluídas as vacinas de ADN e as vacinas de ARNm) que foron aprobadas para a súa comercialización.
1. Terapia xénica in vitro
(1) Strimvelis
Empresa: Desenvolvido por GlaxoSmithKline (GSK).
Time to market: foi aprobado para a súa comercialización pola Unión Europea en maio de 2016.
Indicacións: para o tratamento da inmunodeficiencia combinada grave (SCID).
Observacións: o proceso xeral desta terapia consiste en obter primeiro as propias células nai hematopoéticas do paciente, expandilas e cultivalas in vitro, despois usar retrovirus para introducir unha copia funcional do xene ADA (adenosina desaminase) nas células nai hematopoéticas e, finalmente, inxectar as células nai hematopoéticas modificadas de novo no corpo.Os resultados clínicos mostran que a taxa de supervivencia a 3 anos dos pacientes con ADA-SCID tratados con Strimvelis é do 100%.
(2) Zalmoxis
Empresa: Producida por Italia MolMed Company.
Tempo de comercialización: obtivo a autorización de comercialización condicional da Unión Europea en 2016.
Indicacións: úsase para a terapia adyuvante do sistema inmunitario dos pacientes despois do transplante de células nai hematopoyéticas.
Observacións: Zalmoxis é unha inmunoterapia xenética suicida de células T aloxénica modificada por vectores retrovirais.Este método utiliza vectores retrovirais para modificar xeneticamente as células T aloxénicas, de xeito que as células T modificadas xeneticamente expresan os xenes suicidas 1NGFR e HSV-TK Mut2 permiten que as persoas usen fármacos de ganciclovir (ganciclovir) en calquera momento para matar as células T que causan reaccións inmunes adversas, previr o posible deterioro da GVHD e proporcionar unha función inmunolóxica postoperatoria para os pacientes con TCHS.
(3) Invossa-K
Empresa: Desenvolvido por TissueGene (KolonTissueGene).
Tempo de comercialización: aprobado para cotizar en Corea do Sur en xullo de 2017.
Indicacións: Para o tratamento da artrite dexenerativa do xeonllo.
Observacións: Invossa-K é unha terapia xénica celular aloxénica que inclúe condrocitos humanos.As células aloxénicas son modificadas xeneticamente in vitro, e as células modificadas poden expresar e segregar factor de crecemento transformante β1 (TGF-β1) despois da inxección intraarticular.β1), mellorando así os síntomas da artrose.Os resultados clínicos mostran que Invossa-K pode mellorar significativamente a artrite do xeonllo.Foi revogado en 2019 pola Administración de Drogas e Alimentos de Corea porque o fabricante etiquetou mal os ingredientes utilizados.
(4) Zynteglo
Empresa: Investigada e desenvolvida por bluebird bio.
Time to market: aprobado pola Unión Europea para a súa comercialización en 2019 e aprobado pola FDA para a súa comercialización nos Estados Unidos en agosto de 2022.
Indicacións: para o tratamento da β-talasemia dependente da transfusión.
Observacións: Zynteglo é unha terapia xénica lentiviral in vitro que introduce unha copia funcional do xene normal da β-globina (xene βA-T87Q-globina) en células nai hematopoyéticas tomadas do paciente a través dun vector lentiviral e, a continuación, reinfunde estas células nai hematopoyéticas autólogas modificadas xeneticamente.Unha vez que o paciente ten un xene normal de βA-T87Q-globina, pode producir proteína HbAT87Q normal, que pode reducir ou eliminar eficazmente a necesidade de transfusión de sangue.É unha terapia única deseñada para substituír transfusións de sangue e medicamentos de por vida para pacientes de 12 anos ou máis.
(5) Skysona
Empresa: Investigada e desenvolvida por bluebird bio.
Time to market: aprobado pola Unión Europea para a súa comercialización en xullo de 2021 e aprobado pola FDA para a súa comercialización nos Estados Unidos en setembro de 2022.
Indicacións: para o tratamento da adrenoleucodistrofia cerebral precoz (CALD).
Observacións: a terapia xénica Skysona é a única terapia xénica única aprobada para o tratamento da adrenoleucodistrofia cerebral en fase inicial (CALD).Skysona (elivaldogene autotemcel, Lenti-D) é un lentiviral de células nai hematopoyéticas de terapia xénica in vitro Lenti-D.O procedemento xeral da terapia é o seguinte: as células nai hematopoyéticas autólogas son extraídas do paciente, transducidas e modificadas por lentivirus que portan o xene humano ABCD1 in vitro e despois reinfundidas ao paciente.Utilízase para tratar pacientes menores de 18 anos, portadores de mutacións no xene ABCD1 e CALD.
(6) Kymriah
EMPRESA: Desenvolvido por Novartis.
Time to market: aprobado pola FDA para comercialización en agosto de 2017.
Indicacións: Tratamento da leucemia linfoblástica aguda de células B (LLA) precursora e do DLBCL en recaída e refractario.
Observacións: Kymriah é un fármaco lentiviral de terapia xénica in vitro, a primeira terapia CAR-T aprobada para a súa comercialización no mundo, dirixida a CD19 e que utiliza un factor coestimulador 4-1BB.Ten un prezo de 475.000 dólares nos Estados Unidos e 313.000 dólares en Xapón.
(7) Yescarta
Empresa: Desenvolvido por Kite Pharma, unha subsidiaria de Gilead (GILD).
Time to market: aprobado pola FDA para comercialización en outubro de 2017;Fosun Kite introduciu a tecnoloxía Yescarta de Kite Pharma e produciua en China despois de obter a autorización.Aprobado para listar no país.
Indicacións: Para o tratamento do linfoma de células B grandes en recaída ou refractario.
Observacións: Yescarta é unha terapia xénica retroviral in vitro, que é a segunda terapia CAR-T aprobada no mundo.Diríxese ao CD19 e adopta o coestimulador do CD28.Ten un prezo de 373.000 dólares nos Estados Unidos.
(8) Tecartus
Empresa: Desenvolvido por Gilead (GILD).
Tempo de comercialización: aprobado pola FDA para comercialización en xullo de 2020.
Indicacións: Para linfoma de células do manto en recaída ou refractario.
Observacións: Tecartus é unha terapia con células CAR-T autóloga dirixida a CD19, e é a terceira terapia CAR-T aprobada para a súa comercialización no mundo.
(9) Breyanzi
EMPRESA: Desenvolvido por Bristol-Myers Squibb (BMS).
Tempo de comercialización: aprobado pola FDA para comercialización en febreiro de 2021.
Indicacións: Linfoma de células B grandes (LBCL) en recaída ou refractario (R/R).
Observacións: Breyanzi é unha terapia xénica in vitro baseada en lentivirus, a cuarta terapia CAR-T aprobada para a comercialización no mundo, dirixida a CD19.A aprobación de Breyanzi é un fito para Bristol-Myers Squibb no campo da inmunoterapia celular, que adquiriu cando adquiriu Celgene por 74.000 millóns de dólares en 2019.
(10) Abecma
Empresa: desenvolvido conjuntamente por Bristol-Myers Squibb (BMS) e bluebird bio.
Tempo de comercialización: aprobado pola FDA para comercialización en marzo de 2021.
Indicacións: mieloma múltiple en recaída ou refractario.
Observacións: Abecma é unha terapia xénica in vitro baseada en lentivirus, a primeira terapia con células CAR-T do mundo dirixida a BCMA e a quinta terapia CAR-T aprobada pola FDA.O principio do fármaco é expresar receptores BCMA quiméricos nas propias células T do paciente mediante a modificación de xenes mediada por lentivirus in vitro.Tratamento para eliminar células T non modificadas xeneticamente en pacientes, e despois reinfundir células T modificadas, que buscan e matan as células cancerosas que expresan BCMA nos pacientes.
(11) Libmeldy
EMPRESA: Desenvolvido por Orchard Therapeutics.
Time to market: aprobado pola Unión Europea para a súa cotización en decembro de 2020.
Indicacións: Para o tratamento da leucodistrofia metacromática (MLD).
Observacións: Libmeldy é unha terapia xénica baseada en células CD34+ autólogas modificadas xeneticamente in vitro por lentivirus.Os datos clínicos mostran que unha única infusión intravenosa de Libmeldy pode alterar eficazmente o curso da MLD de inicio precoz en comparación co deterioro motor e cognitivo grave en pacientes da mesma idade non tratados.
(12) Benoda
Empresa: Desenvolvido por WuXi Giant Nuo.
Tempo de comercialización: aprobado oficialmente pola NMPA en setembro de 2021.
Indicacións: tratamento de pacientes adultos con linfoma de células B grandes en recaída ou refractario (LBCL r/r) despois dunha terapia sistémica de segunda liña ou superior.
Observacións: Beinoda é unha terapia xénica CAR-T anti-CD19 e tamén é o produto principal de WuXi Juro Company.É o segundo produto CAR-T aprobado en China, excepto para o xigante linfoide de células B grandes recaídas/refractarias, o xigante Wuxi, o xigante Wuxi, tamén planea desenvolver a inxección de regiorensai para o tratamento doutras indicacións múltiples, incluído o linfoma folicular (FL), o linfoma de células do manto), o linfoma de blusa crónico (cll), o limfoma do ccelín crónico, o cll. leucemia linfoblástica aguda (todas).
(13) CARVYKTI
Empresa: O primeiro produto de Legend Biotech aprobado para a súa comercialización.
Tempo de comercialización: aprobado pola FDA para comercialización en febreiro de 2022.
Indicacións: para o tratamento do mieloma múltiple en recaída ou refractario (R/R MM).
Observacións: CARVYKTI (ciltacabtagene autoleucel, Cilta-cel para abreviar) é unha terapia xénica inmune de células CAR-T con dous anticorpos dun só dominio dirixidos ao antíxeno de maduración de células B (BCMA).Os datos mostran que CARVYKTI En pacientes con mieloma múltiple en recaída ou refractario que recibiron catro ou máis terapias anteriores (incluíndo inhibidores do proteasoma, inmunomoduladores e anticorpos monoclonais anti-CD38), mostrouse unha taxa de resposta global do 98%.
(14)Ebvallo
EMPRESA: Desenvolvido por Atara Biotherapeutics.
a Comisión Europea (CE) para a súa comercialización en decembro de 2022, é a primeira terapia universal de células T do mundo aprobada para a súa comercialización.
Indicacións: como monoterapia para a enfermidade linfoproliferativa posttransplante (EBV+PTLD) relacionada co virus de Epstein-Barr (EBV), os pacientes que reciben tratamento deben ser adultos e nenos maiores de 2 anos que recibiron previamente polo menos outro medicamento.
Observacións: Ebvallo é unha terapia xénica universal de células T aloxénica específica de EBV que se dirixe e elimina as células infectadas por EBV dun xeito restrinxido por HLA.A aprobación desta terapia baséase nos resultados do estudo de ensaio clínico pivotal de fase 3, e os resultados mostraron que o ORR do grupo HCT e do grupo SOT foi do 50%.A taxa de remisión completa (CR) foi do 26,3%, a taxa de remisión parcial (PR) foi do 23,7% e o tempo medio de remisión (TTR) foi de 1,1 meses.Dos 19 pacientes que lograron a remisión, 11 tiveron unha duración de resposta (DOR) de máis de 6 meses.Ademais, en termos de seguridade, non se produciron reaccións adversas como a enfermidade do enxerto contra o hóspede (GvHD) ou a síndrome de liberación de citocinas relacionada con Ebvallo.
2. Terapia xénica in vivo baseada en vectores virais
(1) Gendicine/Jin Sheng
Empresa: Desenvolvido por Shenzhen Saibainuo Company.
Tempo de comercialización: aprobado para cotizar en China en 2003.
Indicacións: Para o tratamento do carcinoma de células escamosas de cabeza e pescozo.
Nota: Gendicine/Jinyousheng de inxección de adenovirus humano recombinante p53 é un fármaco de terapia xénica con vectores de adenovirus con dereitos de propiedade intelectual independentes propiedade da empresa Shenzhen Saibainuo.O adenovirus humano tipo 5 está composto por adenovirus humano tipo 5. O primeiro é a estrutura principal para o efecto antitumoral do fármaco e o segundo actúa principalmente como portador.O vector de adenovirus transporta o xene terapéutico p53 á célula diana, expresa o xene supresor de tumores p53 na célula diana e a súa expresión xénica. O produto pode regular á alza unha variedade de xenes anticanceríxenos e regular á baixa as actividades dunha variedade de oncoxenes, mellorando así o efecto supresor de tumores do corpo e lograr a finalidade de matar tumores.
(2) Rigvir
Empresa: Desenvolvido por Latima Company, Letonia.
Hora de cotización: Aprobado para cotizar en Letonia en 2004.
Indicacións: Para o tratamento do melanoma.
Observacións: Rigvir é unha terapia xénica baseada nun vector de enterovirus ECHO-7 modificado xeneticamente.Actualmente, a droga foi adoptada en Letonia, Estonia, Polonia, Armenia, Bielorrusia, etc., e tamén está a ser rexistrada pola EMA nos países da UE.Os casos clínicos dos últimos dez anos demostraron que o virus oncolítico Rigvir é seguro e eficaz e pode aumentar a taxa de supervivencia dos pacientes con melanoma entre 4 e 6 veces.Ademais, esta terapia tamén é aplicable a unha variedade de outros tipos de cancro, incluíndo cancro colorrectal, cancro de páncreas, cancro de vexiga, cancro de ril, cancro de próstata, cancro de pulmón, cancro de útero, linfosarcoma, etc.
(3) Oncorina
Empresa: Desenvolvido por Shanghai Sanwei Biological Company.
Tempo de comercialización: aprobado para cotizar en China en 2005.
Indicacións: tratamento de tumores de cabeza e pescozo, cancro de fígado, cancro de páncreas, cancro cervical e outros cancros.
Observacións: Oncorine (安科瑞) é un produto de terapia xénica do virus oncolítico que utiliza adenovirus como portador.Obtense un adenovirus oncolítico, que pode replicarse especificamente en tumores anormais ou deficientes no xene p53, o que leva á lise das células tumorais, matando así as células tumorais.sen danar as células normais.Estudos clínicos demostraron que Ankerui ten unha boa seguridade e eficacia para unha variedade de tumores malignos.
(4) Glybera
Empresa: Desenvolvido por uniQure.
Time to market: aprobado para a súa cotización en Europa en 2012.
Indicacións: Tratamento da deficiencia de lipoproteína lipasa (LPLD) con episodios graves ou recorrentes de pancreatite a pesar dunha dieta restrinxida en graxas.
Observacións: Glybera (alipogene tiparvovec) é un fármaco de terapia xénica baseado en AAV, que usa AAV como portador para transducir o xene terapéutico LPL nas células musculares, para que as células correspondentes poidan producir unha certa cantidade de lipoproteína lipase.A droga foi retirada do mercado en 2017. O motivo da súa retirada pode estar relacionado con dous factores: prezos elevados e demanda limitada do mercado.O custo medio do tratamento do medicamento é de 1 millón de dólares e só un paciente comprou e utilizouno ata agora.Aínda que a compañía de seguros médicos reembolsou 900.000 dólares por iso, tamén supón unha carga relativamente grande para a compañía de seguros.Ademais, as indicacións dirixidas ao medicamento son demasiado raras, cunha taxa de incidencia de aproximadamente 1 en 1 millón e unha alta taxa de diagnóstico erróneo.
(5) Imlíxico
Empresa: Desenvolvido por Amgen.
Tempo de comercialización: en 2015 aprobouse para cotizar nos Estados Unidos e na Unión Europea.
Indicacións: Tratamento de lesións de melanoma que non se poden eliminar completamente mediante cirurxía.
Observacións: Imlygic é un virus do herpes simplex atenuado tipo 1 que foi modificado pola tecnoloxía xenética (eliminando os seus fragmentos de xenes ICP34.5 e ICP47 e inserindo o xene GM-CSF do factor estimulante de colonias de macrófagos de granulocitos humanos no virus) (HSV-1) o virus oncolítico aprobado pola FDA é o primeiro virus xenético aprobado pola FDA.O método de administración é a inxección intralesional, que se pode inxectar directamente nas lesións de melanoma para provocar a ruptura das células tumorais, liberar antíxenos derivados do tumor e GM-CSF e promover respostas inmunitarias antitumorales.
(6) Luxturna
Empresa: Desenvolvido por Spark Therapeutics, unha subsidiaria de Roche.
Tempo de comercialización: foi aprobado para a súa comercialización pola FDA en 2017, e despois aprobado para a súa comercialización en Europa en 2018.
Indicacións: Para o tratamento de nenos e adultos que perderon a visión debido a mutacións do xene RPE65 en dobre copia pero que conservan un número suficiente de células da retina viables.
Observacións: Luxturna é unha terapia xénica baseada en AAV administrada por inxección subretiniana.A terapia xénica utiliza AAV2 como portador para introducir unha copia funcional do xene RPE65 normal nas células da retina do paciente, de xeito que as células correspondentes expresen a proteína RPE65 normal, compensando a deficiencia de proteína RPE65 do paciente, mellorando así a visión do paciente.
(7) Zolgensma
Empresa: Desenvolvido por AveXis, unha subsidiaria de Novartis.
Time to market: aprobado pola FDA para comercialización en maio de 2019.
Indicacións: tratamento de pacientes con atrofia muscular espinal (SMA) menores de 2 anos.
Observacións: Zolgensma é unha terapia xénica baseada no vector AAV.Este medicamento é o único plan de tratamento único para a atrofia muscular espinal aprobado para a súa comercialización no mundo.O lanzamento do medicamento abre unha nova era no tratamento da atrofia muscular espiñal.páxina, é un progreso fito.Esta terapia xénica utiliza o vector scAAV9 para introducir o xene SMN1 normal no paciente mediante infusión intravenosa para producir proteína SMN1 normal, mellorando así a función das células afectadas, como as neuronas motoras.Pola contra, os medicamentos SMA Spinraza e Evrysdi requiren dosificacións repetidas a longo prazo.Spinraza dáse por inxección espinal cada catro meses, e Evrysdi é unha droga oral diaria.
(8) Delytact
Empresa: Desenvolvido por Daiichi Sankyo Company Limited (TYO: 4568).
Tempo de comercialización: aprobación condicional do Ministerio de Sanidade, Traballo e Benestar (MHLW) de Xapón en xuño de 2021.
Indicacións: Para o tratamento do glioma maligno.
Observacións: Delytact é o cuarto produto de terapia xénica do virus oncolítico aprobado a nivel mundial e o primeiro produto do virus oncolítico aprobado para o tratamento do glioma maligno.Delytact é un virus oncolítico do virus herpes simplex tipo 1 (HSV-1) desenvolvido polo doutor Todo e colegas.Delytact introduce mutacións adicionais de deleción no xenoma G207 do HSV-1 de segunda xeración, mellorando a súa replicación selectiva nas células cancerosas e a indución de respostas inmunitarias antitumorales mantendo unha alta seguridade.Delytact é o primeiro HSV-1 oncolítico de terceira xeración que está en fase de avaliación clínica.A aprobación de Delytact en Xapón baséase principalmente nun ensaio clínico de fase 2 dun só brazo.En pacientes con glioblastoma recorrente, Delytact alcanzou o punto final principal da taxa de supervivencia a un ano, e os resultados mostraron que Delytact mostrou unha mellor eficacia en comparación co G207.Forte forza replicativa e maior actividade antitumoral.Isto foi eficaz en modelos de tumores sólidos de mama, próstata, schwannomas, cancro nasofarínxeo, hepatocelular, colorrectal, tumores malignos de vaina nerviosa periférica e cancro de tiroide.
(9) Upstaza
EMPRESA: Desenvolvido por PTC Therapeutics, Inc. (NASDAQ: PTCT).
Time to market: aprobado pola Unión Europea para a súa comercialización en xullo de 2022.
Indicacións: para a deficiencia de L-aminoácido descarboxilase aromático (AADC), está aprobado para o tratamento de pacientes de 18 meses ou máis.
Observacións: Upstaza™ (eladocagene exuparvovec) é unha terapia xénica in vivo con virus adeno-asociado tipo 2 (AAV2) como portador.Os pacientes enferman debido a mutacións no xene que codifica o encima AADC.AAV2 leva un xene san que codifica o encima AADC.A forma de compensación xenética consegue un efecto terapéutico.En teoría, unha administración é eficaz durante moito tempo.É a primeira terapia xénica comercializada que se inxecta directamente no cerebro.A autorización de comercialización é aplicable aos 27 estados membros da UE, así como a Islandia, Noruega e Liechtenstein.
(10) Roctaviano
Empresa: Desenvolvido por BioMarin Pharmaceutical (BioMarin).
Time to market: aprobado para a súa comercialización pola Unión Europea en agosto de 2022;autorización de comercialización da Administración de Medicamentos e Produtos Sanitarios do Reino Unido (MHRA) en novembro de 2022.
Indicacións: Para o tratamento de pacientes adultos con hemofilia A grave que non teñen antecedentes de inhibición do factor FVIII e son negativos para anticorpos AAV5.
Observacións: Roctavian (valoctocoxene roxaparvovec) usa AAV5 como vector e usa o promotor específico do fígado humano HLP para impulsar a expresión do factor VIII de coagulación humano (FVIII) co dominio B eliminado.A decisión da Comisión Europea de aprobar a comercialización do valoctocoxene roxaparvovec baséase nos datos xerais do proxecto de desenvolvemento clínico do medicamento.Entre eles, os resultados do ensaio clínico de fase III GENER8-1 mostraron que, en comparación cos datos do ano anterior á inscrición, despois dunha única infusión de valoctocoxene roxaparvovec, a taxa de hemorraxia anual do suxeito (ABR) redúcese significativamente, a frecuencia de uso do factor VIII de coagulación recombinante (F8) ou a actividade de proteínas no sangue redúcese significativamente no organismo F8.Despois de 4 semanas de tratamento, a taxa anual de uso de F8 do suxeito e o ABR que requiriu tratamento reducíronse nun 99% e nun 84%, respectivamente, e a diferenza foi estatisticamente significativa (p<0,001).O perfil de seguridade foi bo e ningún suxeito experimentou efectos secundarios de inhibición do factor F8, neoplasias ou tromboses, e non se informou de eventos adversos graves (SAE) relacionados co tratamento.
(11) Hemgenix
Empresa: Desenvolvido por UniQure Corporation.
Tempo de comercialización: aprobado pola FDA para comercialización en novembro de 2022.
Indicacións: para o tratamento de pacientes adultos con hemofilia B.
Observacións: Hemgenix é unha terapia xénica baseada no vector AAV5.O medicamento está equipado coa variante xenética do factor de coagulación IX (FIX) FIX-Padua, que se administra por vía intravenosa.Despois da administración, o xene pode expresar o factor de coagulación FIX no fígado e segregar. Despois de entrar no sangue para exercer a función de coagulación, para acadar o propósito do tratamento, teoricamente, unha administración é eficaz durante moito tempo.
(12) Adstiladrin
Empresa: Desenvolvido por Ferring Pharmaceuticals.
Tempo de comercialización: aprobado pola FDA para comercialización en decembro de 2022.
Indicacións: Para o tratamento do cancro de vexiga non muscular invasivo de alto risco (NMIBC) que non responde ao Bacillus Calmette-Guerin (BCG).
Observacións: a adstiladrina é unha terapia xénica baseada nun vector adenoviral non replicante, que pode sobreexpresar a proteína interferón alfa-2b nas células diana, e adminístrase a través dun catéter urinario na vexiga (administrado unha vez cada tres meses), o vector do virus pode infectar eficazmente as células da vexiga e, a continuación, interferir a parede da vexiga e, a continuación, sobreexpresar a proteína e, a continuación, exprimir a parede da vexiga.Este novo método de terapia xénica transforma así as propias células da parede da vexiga do paciente nunha "fábrica" en miniatura que produce interferón, mellorando así a capacidade do paciente para loitar contra o cancro.
A seguridade e a eficacia de Adstiladrin avaliáronse nun estudo clínico multicéntrico que incluíu 157 pacientes con NMIBC de alto risco que non responden a BCG.Os pacientes recibiron Adstiladrin cada tres meses durante ata 12 meses, ou ata unha toxicidade inaceptable para o tratamento ou a reaparición de NMIBC de alto grao.En xeral, o 51 por cento dos pacientes inscritos tratados con Adstiladrin acadaron unha resposta completa (desaparición de todos os signos de cancro observados na cistoscopia, tecido de biopsia e orina).
3. Pequenos fármacos de ácido nucleico
(1) Vitravene
Empresa: desenvolvida conxuntamente por Ionis Pharma (anteriormente Isis Pharma) e Novartis.
Tempo de comercialización: en 1998 e 1999, foi aprobado para a súa comercialización pola FDA e a UE EMA.
Indicacións: para o tratamento da retinite por citomegalovirus en pacientes VIH positivos.
Observacións: Vitravene é un oligonucleótido antisentido, que é o primeiro oligonucleótido aprobado para a súa comercialización no mundo.Na fase inicial de cotización, a demanda do mercado de medicamentos anti-CMV era moi urxente;máis tarde, debido ao desenvolvemento da terapia antirretroviral altamente activa, o número de casos de CMV descendeu drasticamente.Debido á escasa demanda do mercado, a droga lanzouse en 2002 e 2006. Retirada nos países da UE e nos Estados Unidos.
(2) Macugen
Empresa: desenvolvido conjuntamente por Pfizer e Eyetech.
Time to market: aprobado para a súa cotización nos Estados Unidos en 2004.
Indicacións: Para o tratamento da dexeneración macular neovascular relacionada coa idade.
Observacións: Macugen é un fármaco oligonucleótido modificado pexilado, que pode dirixirse e unirse ao factor de crecemento endotelial vascular (subtipo VEGF165), e o método de administración é a inxección intravítrea.
(3) Defitelio
Empresa: Desenvolvido por Jazz Pharmaceuticals.
Tempo de comercialización: foi aprobado para a súa comercialización pola Unión Europea en 2013 e aprobada pola FDA para a súa comercialización en marzo de 2016.
Indicacións: Para o tratamento da enfermidade veno-oclusiva hepática asociada á disfunción renal ou pulmonar despois do transplante de células nai hematopoyéticas.
Observacións: Defitelio é un fármaco oligonucleótido, que é unha mestura de oligonucleótidos con propiedades de plasmina.Retirada do mercado en 2009 por motivos comerciais.
(4) Kynamro
Empresa: desenvolvido conjuntamente por Ionis Pharma e Kastle.
Tempo de comercialización: en 2013, foi aprobado para a súa comercialización nos Estados Unidos como medicamento orfo.
Indicacións: para o tratamento coadyuvante da hipercolesterolemia familiar homocigota.
Observacións: Kynamro é un fármaco oligonucleótido antisentido, que é un oligonucleótido antisentido dirixido ao ARNm do apo B-100 humano.Kynamro adminístrase como 200 mg por vía subcutánea unha vez por semana.
(5) Spinraza
Empresa: Desenvolvido por Ionis Pharmaceuticals.
Time to market: aprobado pola FDA para comercialización en decembro de 2016.
Indicacións: Para o tratamento da atrofia muscular espinal (AME).
Observacións: Spinraza (nusinersen) é un oligonucleótido antisentido.Ao unirse ao sitio de escisión do exón 7 de SMN2, Spinraza pode cambiar a escisión do ARN do xene SMN2, aumentando así a produción de proteína SMN totalmente funcional.En agosto de 2016, BIOGEN exerceu a súa opción de adquirir os dereitos globais de Spinraza.Spinraza só comezou o seu primeiro ensaio clínico en humanos en 2011. En só 5 anos, foi aprobado pola FDA para a súa comercialización en 2016, o que reflicte o pleno recoñecemento da FDA da súa eficacia.O medicamento foi aprobado para a súa comercialización en China en abril de 2019. O ciclo completo de aprobación de Spinraza en China foi de menos de 6 meses, e pasaron 2 anos e 2 meses desde que Spinraza se aprobou por primeira vez nos Estados Unidos.A velocidade de cotización en China xa é moi rápida.Isto tamén débese ao feito de que o 1 de novembro de 2018 o Centro de Avaliación de Medicamentos emitiu o "Aviso sobre a publicación da lista do primeiro lote de novos medicamentos no estranxeiro necesarios con urxencia na práctica clínica" o 1 de novembro de 2018 e incluíuse no primeiro lote de 40 novos medicamentos estranxeiros para a súa revisión acelerada, entre os que se clasificou Spinraza.
(6) Exondys 51
Empresa: Desenvolvido por AVI BioPharma (máis tarde rebautizado como Sarepta Therapeutics).
Tempo de comercialización: en setembro de 2016, foi aprobado para a súa comercialización pola FDA.
Indicacións: para o tratamento da distrofia muscular de Duchenne (DMD) con mutación do xene que omite o exón 51 no xene da DMD.
Observacións: Exondys 51 é un fármaco oligonucleótido antisentido, o oligonucleótido antisentido pode unirse á posición do exón 51 do pre-ARNm do xene DMD, o que resulta na formación de ARNm maduro, parte do exón 51 está bloqueado. mellorando os síntomas do paciente.
(7) Tegsedi
Empresa: Desenvolvido por Ionis Pharmaceuticals.
Time to market: foi aprobado para a súa comercialización pola Unión Europea en xullo de 2018.
Indicacións: Para o tratamento da amiloidose da transtiretina hereditaria (hATTR).
Observacións: Tegsedi é un fármaco oligonucleótido antisentido dirixido ao ARNm da transtiretina.É o primeiro fármaco aprobado no mundo para o tratamento da hATTR.Adminístrase mediante inxección subcutánea.O fármaco reduce a produción de proteína ATTR ao dirixirse ao ARNm da transtiretina (ATTR), e ten unha boa relación beneficio-risco no tratamento de ATTR, e a neuropatía e a calidade de vida do paciente melloráronse substancialmente, e é compatible cos tipos de mutación de TTR. Nin o estadio da enfermidade nin a presenza de miocardiopatía foron relevantes.
(8) Onpattro
Empresa: desenvolvida conxuntamente por Alnylam Corporation e Sanofi Corporation.
Tempo de comercialización: aprobado para a súa cotización nos Estados Unidos en 2018.
Indicacións: Para o tratamento da amiloidose da transtiretina hereditaria (hATTR).
Observacións: Onpattro é un fármaco siRNA dirixido ao ARNm da transtiretina, que reduce a produción de proteína ATTR no fígado e reduce a acumulación de depósitos de amiloide nos nervios periféricos ao dirixirse ao ARNm da transtiretina (ATTR), mellorando e aliviando así os síntomas da enfermidade.
(9) Givlaari
Empresa: Desenvolvido por Alnylam Corporation.
Tempo de comercialización: aprobado pola FDA para comercialización en novembro de 2019.
Indicacións: para o tratamento da porfiria hepática aguda (AHP) en adultos.
Observacións: Givlaari é un fármaco siRNA, que é o segundo fármaco siRNA aprobado para a súa comercialización despois de Onpattro.A forma de administración é a inxección subcutánea.O fármaco diríxese ao ARNm da proteína ALAS1, e o tratamento mensual con Givlaari pode reducir significativa e permanentemente o nivel de ALAS1 no fígado, reducindo así os niveis de ALA neurotóxico e PBG ao rango normal, aliviando así os síntomas da enfermidade do paciente.Os datos mostraron que os pacientes tratados con Givlaari tiveron unha redución do 74% no número de convulsións en comparación co grupo placebo.
(10) Vyondys53
EMPRESA: Desenvolvido por Sarepta Therapeutics.
Tempo de comercialización: aprobado pola FDA para comercialización en decembro de 2019.
Indicacións: para o tratamento de pacientes con DMD con mutación de empalme do exón 53 do xene da distrofina.
Observacións: Vyondys 53 é un fármaco oligonucleótido antisentido, que se dirixe ao proceso de empalme do pre-ARNm da distrofina.O exón 53 está parcialmente truncado, é dicir, non está presente no ARNm maduro, e está deseñado para producir unha distrofina truncada pero aínda funcional, mellorando así a capacidade de exercicio dos pacientes.
(11) Waylivra
Empresa: Desenvolvido por Ionis Pharmaceuticals e a súa filial Akcea Therapeutics.
Tempo de comercialización: foi aprobado para a súa comercialización pola Axencia Europea de Medicamentos (EMA) en maio de 2019.
Indicacións: Como terapia adyuvante ademais do control da dieta en pacientes adultos con síndrome de quilomicronemia familiar (FCS).
Observacións: Waylivra é un oligonucleótido antisentido, que é o primeiro fármaco aprobado para a súa comercialización no mundo para o tratamento da FCS.
(12) Leqvio
Empresa: Desenvolvido por Novartis.
Time to market: aprobado pola Unión Europea para a súa comercialización en decembro de 2020.
Indicacións: Para o tratamento de adultos con hipercolesterolemia primaria (familiar heterocigota e non familiar) ou dislipidemia mixta.
Observacións: Leqvio é un fármaco siRNA dirixido ao ARNm de PCSK9.É a primeira terapia de siRNA do mundo para baixar o colesterol (LDL-C).Adminístrase mediante inxección subcutánea.A droga reduce o nivel de proteína PCSK9 a través da interferencia do ARN, reducindo así o nivel de LDL-C.Os datos clínicos mostran que para os pacientes que non poden reducir os niveis de LDL-C ao nivel obxectivo despois do tratamento coa dose máxima tolerada de estatinas, Leqvio pode reducir o LDL-C nun 50%.
(13)Oxlumo
Empresa: Desenvolvido por Alnylam Pharmaceuticals.
Time to market: aprobado pola Unión Europea para a súa comercialización en novembro de 2020.
Indicacións: Para o tratamento da hiperoxaluria primaria tipo 1 (PH1).
Observacións: Oxlumo é un fármaco siRNA dirixido ao ARNm da hidroxiácido oxidase 1 (HAO1) e o método de administración é a inxección subcutánea.O fármaco foi desenvolvido usando a última tecnoloxía de estabilización mellorada de Alnylam, a tecnoloxía de conxugación ESC-GalNAc, que permite a administración subcutánea de siRNA cunha maior persistencia e potencia.O fármaco degrada ou inhibe o ARNm da hidroxiácido oxidase 1 (HAO1), reduce o nivel de glicolato oxidase no fígado e despois consome o substrato necesario para a produción de oxalato, reducindo a produción de oxalato para controlar a progresión da enfermidade nos pacientes e mellorar os síntomas da enfermidade.
(14) Viltepso
Empresa: Desenvolvido por NS Pharma, unha subsidiaria de Nippon Shinyaku.
Tempo de comercialización: aprobado pola FDA para comercialización en agosto de 2020.
Indicacións: para o tratamento da distrofia muscular de Duchenne (DMD) con mutación do xene que omite o exón 53 no xene da DMD.
Observacións: Viltepso é un fármaco oligonucleótido antisentido que pode unirse á posición do exón 53 do pre-ARNm do xene DMD, o que fai que parte do exón 53 sexa extirpado despois da formación de ARNm maduro, corrixindo así parcialmente o marco de lectura do ARNm.
(15) Amondys 45
Empresa: Desenvolvido por Sarepta Therapeutics.
Tempo de comercialización: aprobado pola FDA para comercialización en febreiro de 2021.
Indicacións: para o tratamento da distrofia muscular de Duchenne (DMD) con mutación do xene que omite o exón 45 no xene da DMD.
Observacións: Amondys 45 é un fármaco oligonucleótido antisentido, o oligonucleótido antisentido pode unirse á posición do exón 45 do pre-ARNm do xene DMD, o que provoca que a parte do exón 45 se bloquee despois da formación de ARNm maduro. mellorando os síntomas do paciente.
(16) Amvuttra (vutrisiran)
Empresa: Desenvolvido por Alnylam Pharmaceuticals.
Tempo de comercialización: aprobado pola FDA para comercialización en xuño de 2022.
Indicacións: para o tratamento da amiloidose da transtiretina hereditaria con polineuropatía (hATTR-PN) en adultos.
Observacións: Amvuttra (Vutrisiran) é un fármaco siRNA dirixido ao ARNm da transtiretina (ATTR), administrado por inxección subcutánea.Vutrisiran baséase no deseño da plataforma de entrega de conxugados de química de estabilidade mellorada (ESC)-GalNAc de Alnylam cunha potencia aumentada e estabilidade metabólica.A aprobación da terapia baséase nos datos de 9 meses do seu estudo clínico de Fase III (HELIOS-A), e os resultados globais mostran que a terapia mellorou os síntomas da hATTR-PN e que máis do 50% do estado dos pacientes reverteuse ou deixou de empeorar.
4. Outros fármacos de terapia xénica
(1) Rexin-G
Empresa: Desenvolvido por Epeius Biotech.
Tempo de comercialización: en 2005, foi aprobado para a súa comercialización pola Administración de Drogas e Alimentos de Filipinas (BFAD).
Indicacións: Para o tratamento de cancros avanzados resistentes á quimioterapia.
Observacións: Rexin-G é unha inxección de nanopartículas cargadas con xenes.Introduce o xene mutante da ciclina G1 nas células diana a través dun vector retroviral para matar específicamente os tumores sólidos.O método de administración é a infusión intravenosa.Como un fármaco dirixido ao tumor que busca e destrúe activamente as células cancerosas metastásicas, ten un certo efecto curativo en pacientes que fallaron con outros fármacos contra o cancro, incluídos os biolóxicos dirixidos.
(2) Neovasculgen
Empresa: Desenvolvido polo Human Stem Cells Institute.
Hora de cotización: aprobouse para a súa inclusión en Rusia o 7 de decembro de 2011 e despois lanzouse en Ucraína en 2013.
Indicacións: para o tratamento da enfermidade arterial vascular periférica, incluída a isquemia grave dos membros.
Observacións: Neovasculgen é unha terapia xénica baseada en plásmidos de ADN.O xene do factor de crecemento endotelial vascular (VEGF) 165 constrúese na columna vertebral do plásmido e infúndese aos pacientes.
(3) Colatexeno
Empresa: desenvolvida conxuntamente pola Universidade de Osaka e empresas de capital risco.
Time to market: aprobado polo Ministerio de Sanidade, Traballo e Benestar de Xapón en agosto de 2019.
Indicacións: Tratamento da isquemia crítica das extremidades inferiores.
Observacións: Collategene é unha terapia xénica baseada en plásmidos, o primeiro fármaco de terapia xénica doméstica producido por AnGes, unha empresa de terapia xénica en Xapón.O compoñente principal deste fármaco é un plásmido espido que contén a secuencia do xene do factor de crecemento do hepatocito humano (HGF).Se a droga se inxecta nos músculos dos membros inferiores, o HGF expresado promoverá a formación de novos vasos sanguíneos ao redor dos vasos sanguíneos ocluídos.Os ensaios clínicos confirmaron o seu efecto na mellora das úlceras.
Como Foregene pode axudar ao desenvolvemento da terapia xénica?
Axudamos a aforrar o tempo de cribado no cribado a gran escala, na fase inicial do desenvolvemento de fármacos siRNA.
Máis detalles visita:
https://www.foreivd.com/cell-direct-rt-qpcr-kit-direct-rt-qpcr-series/
Hora de publicación: 27-12-2022



