



- A PCR é un método usado para amplificar o ADN a partir dunha pequena cantidade de molde de ADN.A RT-PCR usa a transcrición inversa para producir un molde de ADN a partir dunha fonte de ARN que despois pode ser amplificado.
- A PCR e a RT-PCR son normalmente reaccións finais, mentres que a qPCR e a RT-qPCR usan a cinética da velocidade de síntese do produto durante a reacción da PCR para cuantificar a cantidade de molde presente.
- Os métodos máis novos, como a PCR dixital, proporcionan a cuantificación absoluta do molde inicial de ADN, mentres que métodos como a PCR isotérmica reducen a necesidade de equipos caros para proporcionar resultados fiables.
A reacción en cadea da polimerase (PCR) é unha técnica de bioloxía molecular relativamente sinxela e moi utilizada para amplificar e detectar secuencias de ADN e ARN.En comparación cos métodos tradicionais de clonación e amplificación de ADN, que moitas veces poden levar días, a PCR require só unhas horas.A PCR é moi sensible e require un modelo mínimo para a detección e amplificación de secuencias específicas.Os métodos básicos de PCR avanzaron aínda máis desde a simple detección de ADN e ARN.A continuación, ofrecemos unha visión xeral dos diferentes métodos de PCR e dos reactivos que ofrecemos en Enzo Life Sciences para as túas necesidades de investigación.Pretendemos axudar aos científicos a acceder rapidamente aos reactivos de PCR para usar no seu próximo proxecto de investigación.
PCR
Para a PCR estándar, todo o que necesitas é unha ADN polimerase, magnesio, nucleótidos, cebadores, o modelo de ADN a amplificar e un termociclador.O mecanismo de PCR é tan sinxelo coma o seu propósito: 1) o ADN de dobre cadea (dsDNA) é desnaturalizado por calor, 2) os cebadores alíñanse ás cadeas simples de ADN e 3) os cebadores son estendidos pola ADN polimerase, o que dá lugar a dúas copias do ADN. cadea de ADN orixinal.O proceso de desnaturalización, recocido e alongamento nunha serie de temperaturas e tempos coñécese como un ciclo de amplificación (Fig. 1).
|
|
| Figura 1.Representación esquemática dun ciclo de amplificación mediante PCR. |
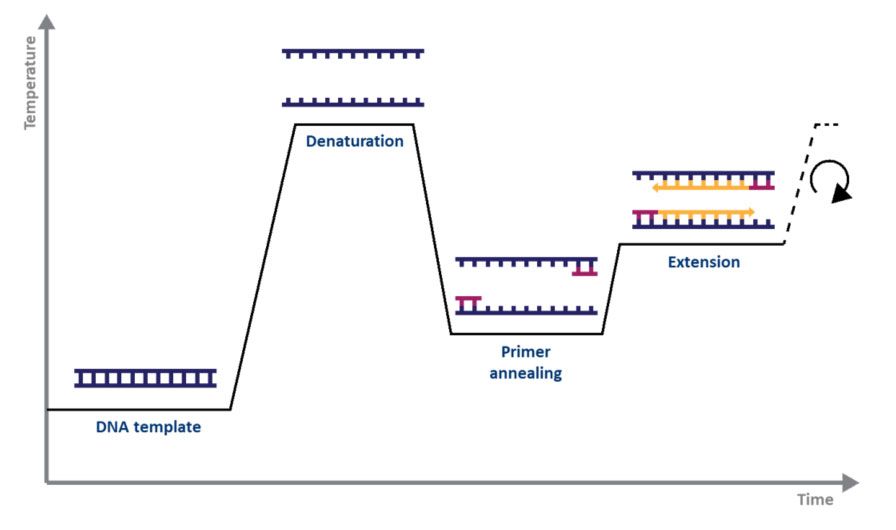
Cada paso do ciclo debe optimizarse para o modelo e o conxunto de imprimacións utilizados.Este ciclo repítese unhas 20-40 veces e despois pódese analizar o produto amplificado, normalmente mediante xel de agarosa (Fig. 2).
| |
| Figura 2.Amplificación dun molde de ADN mediante PCR e análise mediante electroforese en xel de agarosa. |
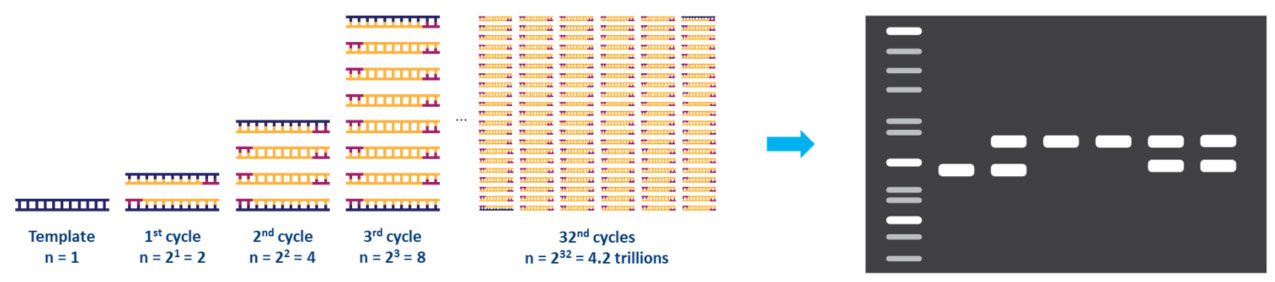
Como a PCR é un método moi sensible e son necesarios volumes moi pequenos para reaccións individuais, recoméndase a preparación dunha mestura principal para varias reaccións.A mestura mestra debe estar ben mesturada e despois dividida polo número de reaccións, garantindo que cada reacción conteña a mesma cantidade de encima, dNTPs e cebadores.Moitos provedores, como Enzo Life Sciences, tamén ofrecen mesturas de PCR que xa o conteñen todo, excepto cebadores e a plantilla de ADN.
As rexións ricas en guanina/citosina (ricos en GC) representan un desafío nas técnicas estándar de PCR.As secuencias ricas en GC son máis estables que as secuencias con menor contido en GC.Ademais, as secuencias ricas en GC tenden a formar estruturas secundarias, como bucles de horquilla.Como resultado, as dobres cadeas ricas en GC son difíciles de separar completamente durante a fase de desnaturalización.En consecuencia, a ADN polimerase non pode sintetizar a nova cadea sen obstáculos.Unha temperatura de desnaturalización máis alta pode mellorar isto, e os axustes cara a unha temperatura de recocido máis alta e un tempo de recocido máis curto poden evitar a unión inespecífica de cebadores ricos en GC.Os reactivos adicionais poden mellorar a amplificación de secuencias ricas en GC.O DMSO, o glicerol e a betaína axudan a interromper as estruturas secundarias que son causadas polas interaccións GC e facilitan así a separación das dobres cadeas.
PCR de inicio en quente
A amplificación inespecífica é un problema que pode ocorrer durante a PCR.A maioría das ADN polimerases que se usan para a PCR funcionan mellor a temperaturas de entre 68 °C e 72 °C.Non obstante, a enzima tamén pode ser activa a temperaturas máis baixas, aínda que en menor grao.A temperaturas moi inferiores á temperatura de recocido, os cebadores poden unirse de forma inespecífica e levar a unha amplificación inespecífica, aínda que a reacción se realice no xeo.Isto pódese evitar empregando inhibidores da polimerase que se disocian da ADN polimerase só unha vez que se alcanza unha determinada temperatura, de aí o termo PCR de inicio en quente.O inhibidor pode ser un anticorpo que se une á polimerase e se desnaturaliza á temperatura de desnaturalización inicial (normalmente 95 °C).
Polimerase de alta fidelidade
Aínda que as ADN polimerases amplifican con bastante precisión ata a secuencia modelo orixinal, pódense producir erros na coincidencia de nucleótidos.As faltas de coincidencia en aplicacións como a clonación poden producir transcricións truncadas e proteínas mal traducidas ou inactivas augas abaixo.Para evitar estes desaxustes, identificáronse e incorporáronse ao fluxo de traballo polimerases con actividade de "corrección".A primeira polimerase de corrección de probas, Pfu, identificouse en 1991 en Pyrococcus furiosus.Este encima Pfu ten unha actividade exonuclease de 3' a 5'.A medida que o ADN se amplifica, a exonuclease elimina os nucleótidos que non coinciden no extremo 3' da cadea.Despois substitúese o nucleótido correcto e continúa a síntese de ADN.A identificación de secuencias de nucleótidos incorrectas baséase na afinidade de unión polo nucleósido trifosfato correcto co encima, onde a unión ineficiente retarda a síntese e permite a substitución correcta.A actividade de corrección da polimerase Pfu produce menos erros na secuencia final en comparación coa ADN polimerase Taq.Nos últimos anos, identificáronse outras enzimas de corrección de probas e fixéronse modificacións da enzima Pfu orixinal para reducir aínda máis a taxa de erro durante a amplificación do ADN.
RT-PCR
A PCR de transcrición inversa, ou RT-PCR, permite o uso de ARN como molde.Un paso adicional permite a detección e amplificación de ARN.O ARN transcríbese inversamente a ADN complementario (ADNc), mediante a transcriptase inversa.A calidade e pureza do molde de ARN son esenciais para o éxito da RT-PCR.O primeiro paso da RT-PCR é a síntese dun híbrido ADN/ARN.A transcriptase inversa tamén ten unha función RNase H, que degrada a porción de ARN do híbrido.A molécula de ADN monocatenario complétase entón pola actividade da ADN polimerase dependente do ADN da transcriptase inversa en ADNc.A eficiencia da reacción da primeira cadea pode afectar o proceso de amplificación.A partir de aquí, utilízase o procedemento estándar de PCR para amplificar o ADNc.A posibilidade de converter o ARN en ADNc mediante RT-PCR ten moitas vantaxes, e utilízase principalmente para a análise da expresión xénica.O ARN é monocatenario e moi inestable, o que fai que sexa difícil traballar con el.Normalmente serve como primeiro paso na qPCR, que cuantifica as transcricións de ARN nunha mostra biolóxica.
qPCR e RT-qPCR
A PCR cuantitativa (qPCR) úsase para detectar, caracterizar e cuantificar ácidos nucleicos para numerosas aplicacións.Na RT-qPCR, as transcricións de ARN adoitan cuantificarse transcribíndoas primeiro a ADNc, como se describiu anteriormente, e despois lévase a cabo a qPCR.Como na PCR estándar, o ADN amplificase mediante tres pasos que se repiten: desnaturalización, recocido e alongamento.Non obstante, na qPCR, o etiquetado fluorescente permite a recollida de datos a medida que avanza a PCR.Esta técnica ten moitos beneficios debido á variedade de métodos e químicas dispoñibles.
Na qPCR baseada en colorantes (normalmente verde), a etiquetaxe fluorescente permite a cuantificación das moléculas de ADN amplificadas empregando o uso dun colorante de unión a dsDNA.Durante cada ciclo mídese a fluorescencia.O sinal de fluorescencia aumenta proporcionalmente á cantidade de ADN replicado.Polo tanto, o ADN cuantificase en "tempo real" (Fig. 3).As desvantaxes da qPCR baseada en colorantes son que só se pode examinar unha diana á vez e que o colorante unirase a calquera ADN-ds presente na mostra.
|
|
| Figura 3.Amplificación dunha plantilla de ADN mediante qPCR e medición do sinal de fluorescencia en tempo real. |
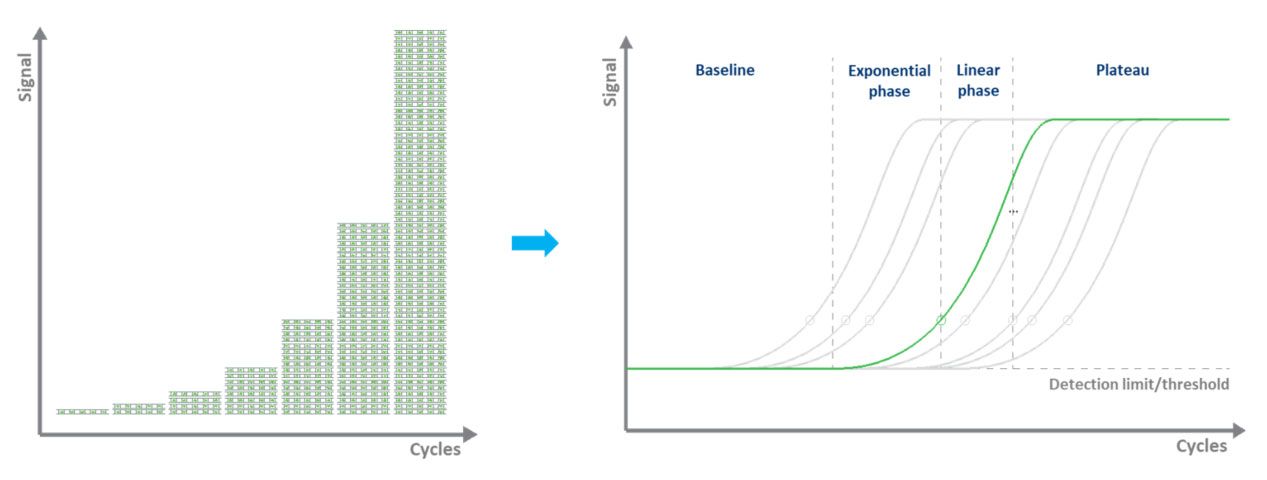
Na qPCR baseada en sondas, pódense detectar simultáneamente moitas dianas en cada mostra, pero isto require a optimización e o deseño dunha(s) sonda(s) específica(s) utilizada(s) ademais dos cebadores.Existen varios tipos de deseños de sondas dispoñibles, pero o tipo máis común é unha sonda de hidrólise, que incorpora un fluoróforo e un extintor.A transferencia de enerxía de resonancia de fluorescencia (FRET) impide a emisión do fluoróforo a través do extintor mentres a sonda está intacta.Non obstante, durante a reacción de PCR, a sonda hidrolízase durante a extensión do cebador e a amplificación da secuencia específica á que está unida.A escisión da sonda separa o fluoróforo do quencher e dá lugar a un aumento da fluorescencia dependente da amplificación (Fig. 4).Así, o sinal de fluorescencia dunha reacción de qPCR baseada en sonda é proporcional á cantidade de secuencia diana da sonda presente na mostra.Dado que a qPCR baseada en sondas é máis específica que a qPCR baseada en colorantes, adoita ser a tecnoloxía utilizada nos ensaios de diagnóstico baseados en qPCR.
| |
| Figura 4.Diferenzas entre a qPCR baseada en colorantes e baseada en sondas. |
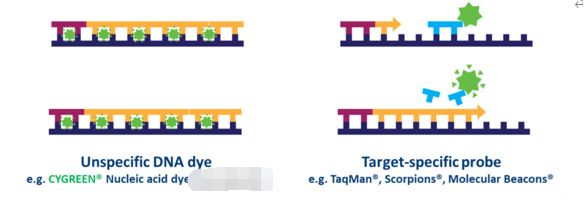
Amplificación isotérmica
As técnicas de PCR mencionadas anteriormente requiren un equipo de termociclado caro para aumentar e baixar con precisión as temperaturas da cámara para os pasos de desnaturalización, recocido e extensión.Desenvolvéronse unha serie de técnicas que non precisan de dispositivos tan precisos e que se poden realizar nun simple baño de auga ou mesmo dentro das células de interese.Estas técnicas denomínanse colectivamente amplificación isotérmica e funcionan baseadas na amplificación exponencial, lineal ou en cascada.
O tipo máis coñecido de amplificación isotérmica é a amplificación isotérmica mediada por bucle, ou LAMP.LAMP utiliza amplificación exponencial a 65⁰C para amplificar o ADN ou o ARN molde.Cando se realiza LAMP, úsanse de catro a seis cebadores complementarios de rexións do ADN diana cunha ADN polimerase para sintetizar novo ADN.Dous destes cebadores teñen secuencias complementarias que recoñecen as secuencias dos outros cebadores e únenas, permitindo que se forme unha estrutura de "bucle" no ADN recén sintetizado que axuda a recozir o cebador nas seguintes roldas de amplificación.A LAMP pode visualizarse mediante múltiples métodos, incluíndo fluorescencia, electroforese en xel de agarosa ou colorimetría.A facilidade para visualizar e detectar a presenza ou ausencia de produto mediante colorimetría e a falta de equipos caros necesarios fixeron de LAMP unha opción adecuada para as probas de SARS-CoV-2 en áreas nas que as probas de laboratorio clínico non estaban facilmente dispoñibles, ou o almacenamento e o transporte de mostras. non era viable, ou en laboratorios que antes non tiñan equipos de termociclado PCR.
Hora de publicación: 19-Ago-2023



